DOI:
10.1039/C5RA26575D
(Paper)
RSC Adv., 2016,
6, 6896-6904
A sensitive method for the determination of gold and palladium based on dispersive liquid–liquid microextraction combined with flame atomic absorption spectrometric determination using N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea
Received
12th December 2015
, Accepted 5th January 2016
First published on 8th January 2016
Abstract
A new method for the determination of gold and palladium was developed by dispersive liquid–liquid microextraction separation–preconcentration and flame atomic absorption spectrometry detection. In the proposed approach, N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea (MPPT) was synthesized as a complexing agent. The complexation ability of the MPPT was explored by examining the effect of a series of heavy metal ions, including Mn2+, Pd2+, Ni2+, Cd2+, Co2+, Cu2+, Au3+, Pb2+, Zn2+ and Fe3+, using the DLLME procedure. The MPPT exhibited pronounced selectivity toward Pd2+ and Au3+ ions at different pH levels. Factors influencing the extraction efficiency and complex formation were examined, i.e. the pH of the sample solution, the concentration of the chelating agent, the extraction and dispersive solvent type and volume, the sample volume, and foreign ions, etc. Optimal conditions for quantitative recoveries were pH 5.5 for gold and pH 1.5 for palladium, 125 μL of % 0.4 MPPT, 1200 μL of methanol and 125 μL of carbon tetrachloride. The presented method showed a good linearity within a range of 30–230 and 25–200 μg L−1 with the detection limits of 1.75 and 1.65 μg L−1 for Au and Pd, respectively. The relative standard deviation (RSD) was below 2.8% at 50 μg L−1 for both ions (n = 10). The developed method was simple, fast, cost efficient, and sensitive for the extraction and preconcentration of gold and palladium in samples of liquids (sea, stream water) and solids (stream sediment, ores, and electronic waste).
1. Introduction
Gold is an important element in our life due to its high ductility, corrosion resistance, and catalytic properties, and is widely used in a variety of industries such as electronics, electroplating, fine-chemicals, and jewellery.1,2 Although it is not a toxic element, experiments regarding allergic eczematous dermatitis have been published.3 Palladium is an element of increasing importance nowadays because of its physical and chemical properties. It is used in as an automotive catalyst as well as in electronics and dentistry.4 Palladium is biologically inactive, however certain palladium compounds have been reported as having potential cytotoxic effects on humans including liver and kidney damage, asthma, allergies, and various other health problems.5 Concentrations of palladium (0.01–0.02 mg L−1) and gold (0.004 mg L−1) in environmental and biological samples are reportedly very low.6,7 Sensitive analytical techniques like ICP-MS8 and GFAAS9 are necessary for determining trace amounts of Pd and Au ions, but are both time-consuming and expensive. FAAS, an alternative, offers covetable characteristics such as low cost in terms of operational facilities.10 However, the determination of trace metal ions in environmental samples is sometimes insufficient due to their low concentrations and the effects of their matrices. Hence, pre-concentration and separation methods are often required first. For the separation and pre-concentration of Pd, several methods exist including coprecipitation,11 liquid–liquid extraction (LLE),12,13 solid-phase extraction (SPE),14,15 cloud point extraction (CPE)16 and hollow fibre liquid phase microextraction (HFLPME).17 These techniques can often be time-consuming and, the use of large organic solvents limit their applications. As a counter to this, there has been growing interest in the development of miniaturized preconcentration methods known as microextraction, which include solid phase microextraction (SPME)18,19 and single drop microextraction (SDME).20 In this context, dispersive liquid–liquid microextraction (DLLME) consists of a miniaturization of the LLE based on the ternary component solvent systems whereby the extraction (μL) and disperser (mL) solvents are rapidly injected into the aqueous sample using a syringe. The formation of a cloudy solution contains fine droplets of extraction solvent fully dispersed into the aqueous phase. After centrifugation, this causes the accumulation of the extraction phase at the bottom of the test tube.21 The DLLME method has gained widespread attention due to its simplicity of operation, rapidity, low cost consumption of organic solvents and reagents, and high recovery and enrichment factors. It has proven to be a suitable preconcentration procedure for palladium and gold ions.22–24
In this study, we synthesized a new N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea ligand and used it in dispersive liquid–liquid microextraction for the preconcentration of Au and Pd ions in environmental samples, at trace levels, using flame atomic absorption spectrometry. The DLLME technique used an appropriate mixture of carbon tetrachloride (extraction solvent), methanol–ethanol (dispersive solvent), and MPPT (chelating agent) for determining gold and palladium ions from solid and aqueous samples. The complex stability constants and complex compositions of Au3+ and Pd2+ cations with the new ligand were examined using spectrophotometric titrations.
2. Experimental
2.1. Apparatus
A Perkin-Elmer A Analyst 400 flame atomic absorption spectrometer (Waltham, United States) in an air/acetylene flame with a 10 cm-long burner head flame atomic absorption spectrophotometer was used. The gold and palladium hollow cathode lamps, operated at 12 mA and 6 mA, were utilized as the radiation source. Measurements were carried out in the peak height mode at 242.80 and 244.79 nm, respectively. A deuterium lamp was used for background correction. The absorption spectra of the compound were recorded using a Specord 210 Plus model spectrophotometer (Jena, Germany). The IR spectra were recorded as potassium bromide pellets using a Perkin-Elmer 1600 series FTIR spectrometer (USA). 1H NMR and 13C NMR spectra were registered in CDCl3 on a BRUKER AVENE III 400 MHz NMR (USA). The mass spectra of the reagent were obtained on an Agilent LC/MS-TOF instrument (USA). The melting points of the synthesized compounds were determined in open capillaries on a Büchi B-540 melting point apparatus. The Milestones Ethos D (Milestore Inc., Italy) with a closed vessel microwave system was standardized for the digestion of the solid samples, and programmable for a pressure of 1450 psi and temperature of 300 °C. A pH meter, the Hanna pH-211model digital glass electrode, was used for measuring pH values of the aqueous phase. A Sigma 3-16P model centrifuge (Sigma laborzentrifugen GmbH, Germany) was employed to centrifuge the solutions. Ultra-pure water, produced by the Merck – Millipore Direct-Q 8UV system (Darmstadt, Germany), was used throughout.
2.2. Reagents
The stock standard solution (1000 mg L−1) of Au and Pd was obtained using the Merck (Darmstadt, Germany). Working standard solutions were obtained by appropriate dilution of the stock standard solution. The chelating agent, N-(6-morpholin-4 ylpyridin-3-yl)-N′-phenylthiourea (MPPT) was synthesized and purified in our laboratories. A 0.4% (w/v) solution of the chelating agent was prepared by dissolving the proper amount of the agent in methanol/dimethylsulfoxide (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Phosphoric acid/sodium dihydrogen phosphate (0.1 mol L−1) was used to adjust the pH 1–3. Ammonium acetate buffers (0.1 mol L−1) were prepared by adding an appropriate amount of acetic acid to ammonium acetate solutions resulting in solutions of pH 4–6. For pH 7, a phosphate (0.1 mol L−1) buffer solution was prepared by adding an appropriate amount of disodium hydrogen phosphate to sodium dihydrogen phosphate. Ammonium chloride buffer solutions (0.1 mol L−1) were used for pH 8–10. 2-Chloro-5-nitropyridine (97%, Sigma-Aldrich), morpholine (99%, Sigma-Aldrich), hydrazine hydrate (100%, Darmstadt, Merck), palladium on carbon (10% Pd, Sigma-Aldrich), and phenylisothiocyanate (98% Acros-Organics, New Jersey, USA) were used for the synthesis of N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea. The remaining used chemicals, including dimethylsulfoxide (analytical), carbon tetrachloride (analytical), chloroform (analytical grade), dichloromethane (analytical grade), carbon disulfide (for spectroscopy), methanol (for spectroscopy), ethanol (for spectroscopy), acetone (suprasolv), acetonitrile (HPLC grade), and tetra hydro furan (suprasolv) were provided by Merck (Darmstadt, Germany). All other reagents used were of the highest available purity and of at least analytical reagent grade. The sandy soil (CRM-SA-C) certified reference material used in the DLLME studies was acquired from High-Purity Standard Inc., USA.
:1). Phosphoric acid/sodium dihydrogen phosphate (0.1 mol L−1) was used to adjust the pH 1–3. Ammonium acetate buffers (0.1 mol L−1) were prepared by adding an appropriate amount of acetic acid to ammonium acetate solutions resulting in solutions of pH 4–6. For pH 7, a phosphate (0.1 mol L−1) buffer solution was prepared by adding an appropriate amount of disodium hydrogen phosphate to sodium dihydrogen phosphate. Ammonium chloride buffer solutions (0.1 mol L−1) were used for pH 8–10. 2-Chloro-5-nitropyridine (97%, Sigma-Aldrich), morpholine (99%, Sigma-Aldrich), hydrazine hydrate (100%, Darmstadt, Merck), palladium on carbon (10% Pd, Sigma-Aldrich), and phenylisothiocyanate (98% Acros-Organics, New Jersey, USA) were used for the synthesis of N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea. The remaining used chemicals, including dimethylsulfoxide (analytical), carbon tetrachloride (analytical), chloroform (analytical grade), dichloromethane (analytical grade), carbon disulfide (for spectroscopy), methanol (for spectroscopy), ethanol (for spectroscopy), acetone (suprasolv), acetonitrile (HPLC grade), and tetra hydro furan (suprasolv) were provided by Merck (Darmstadt, Germany). All other reagents used were of the highest available purity and of at least analytical reagent grade. The sandy soil (CRM-SA-C) certified reference material used in the DLLME studies was acquired from High-Purity Standard Inc., USA.
2.3. Synthesis of N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea (MPPT)
2-Chloro-5-nitropyridin (1.410 g, 0.010 mol) was dissolved in morpholine 1 (0.020 L) and the reaction mixture was stirred and heated under reflux for 8 hours. Afterwards, the reaction mixture was cooled down to room temperature and the product was precipitated. The crude solid product was crystallized from ethanol and obtained a pure compound 2 (yield: 1.920 g, 92%). Then, a mixture of compound 2 (1.920 g, 0.091 mol), palladium-carbon catalyst (0.590 g, 0.005 mmol) and hydrazine hydrate (2.50 mL, 0.050 mmol) in butanol (0.050 L) was heated under reflux for 6 hours, then cooled down to room temperature and the catalyzer filtered off. The solvent was evaporated and the crude product was crystallized from ethanol and obtained pure compound 3 (yield: 1.320 g, 74%). Finally, a solution of compound 3 (1.320 g, 7.3 mmol) and phenylisothiocyanate (0.36 mL, 0.003 mol) in 50 mL of butanol was heated under reflux for 7 hours. At the end of the reaction, the crude product collapsed. The precipitate found was filtered and crystallized from ethanol. This compound was characterized by means of IR, 1H NMR, 13C NMR and TOF-LC/MS spectroscopies. The synthetic pathways to the MPPT are presented in Fig. 1.
 |
| | Fig. 1 The synthesis scheme of the MPPT. | |
IR (KBr pellet, ν cm−1): 3212 (NH), 3171 (NH), 3034 (Ar-CH), 1537 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1234 (CS), 1109 (C–O). 1H NMR (CDCl3, δ ppm): 3.55 (t, 4H, N–2CH2, J = 10.0 Hz), 3.83 (t, 4H, O–2CH2, J = 9.6 Hz), 6.68 (d, 1H, Ar-H, J = 8.8 Hz), 7.28 (s, 1H, CH), 7.39–7.47 (m, 4H, Ar-H), 7.67 (d, 2H, 2CH, J = 8.0 Hz), 7.84 (brs, 1H, NH), 8.11 (d, 1H, NH, J = 2.4 Hz). 13C NMR (CDCl3, δ ppm): 181.19 (CS), ArC: [158.11 (C), 145.59 (CH), 136.93 (C), 136.52 (2CH), 129.72 (CH), 127.23 (CH), 125.34 (2CH), 125.25 (C), 106.61 (CH)], 66.63 (O–2CH2), 45.51 (N–2CH2). TOF-LC/MS (ESI): m/z (%) 315.12 [M + H]+, (100), 337.11 [M + Na]+.
N), 1234 (CS), 1109 (C–O). 1H NMR (CDCl3, δ ppm): 3.55 (t, 4H, N–2CH2, J = 10.0 Hz), 3.83 (t, 4H, O–2CH2, J = 9.6 Hz), 6.68 (d, 1H, Ar-H, J = 8.8 Hz), 7.28 (s, 1H, CH), 7.39–7.47 (m, 4H, Ar-H), 7.67 (d, 2H, 2CH, J = 8.0 Hz), 7.84 (brs, 1H, NH), 8.11 (d, 1H, NH, J = 2.4 Hz). 13C NMR (CDCl3, δ ppm): 181.19 (CS), ArC: [158.11 (C), 145.59 (CH), 136.93 (C), 136.52 (2CH), 129.72 (CH), 127.23 (CH), 125.34 (2CH), 125.25 (C), 106.61 (CH)], 66.63 (O–2CH2), 45.51 (N–2CH2). TOF-LC/MS (ESI): m/z (%) 315.12 [M + H]+, (100), 337.11 [M + Na]+.
2.4. Recommended procedure
The aliquots of 10.0 mL sample solution containing 0.5 μg of Au (pH 5.5) were placed in a 10 mL screw cap glass test tube with a conic bottom. The amount of 1200 μL of methanol (disperser solvent), 125 μL % 0.4 MPPT (chelating agent) and 125 μL of carbon tetrachloride (extraction solvent) was injected rapidly into the sample solution. A cloudy solution (water, ethanol, carbon tetrachloride) was formed in the test tube. In this step, Au reacted with MPPT and was extracted into the fine droplets of carbon tetrachloride. Then, after 5 minutes of standing time, the solution was centrifuged at 3000 rpm for 3 min, and the dispersed fine droplets of carbon tetrachloride were deposited into the bottom of conical test tube. The residue phase was diluted to 0.5 mL with 0.1 mol L−1 HNO3 containing methanol (as dilute solvent) and injected into the FAAS for Au analysis. 10 mL of the solution containing 0.5 μg Pd (pH = 1.5) was transferred into a 15 mL PTFE conical-bottomed tube, and then a mixture of 1200 μL ethanol, 125 μL % 0.4 MPPT and 125 μL carbon tetrachloride was added quickly to the solution. Following that, all the necessary procedures as described above were applied.
2.5. Sample preparation
Microwave digestion was chosen for the decomposing of all solid samples. Solid samples of 0.4 g ore-1 (Akoluk, Ordu, Turkey), 0.4 g ore-2 (Bolkardağ, Niğde, Turkey), 0.4 g ore-3 (Tüprak, Uşak, Turkey) and 0.4 g sediment (Maçka stream, Trabzon, Turkey) were accurately weighed and dissolved into a mixture of HNO3, HCl, HF and H2O2 (3.5:3.5:1:1 volume ratio) into a polytetrafluoroethylene vessel. Electronic waste samples (0.15 g) were weighed and transferred into Teflon vessels, 3.5 mL concentrated of HNO3 and 3.5 mL of HCl were added for the digestion. After the digestion procedure, all the solutions were evaporated to near dryness. Purified water was added to the residues. The suspensions were filtered through a blue band filter paper (Macherey-Nagel, Duren, Germany). The final solutions were completed to 25 mL (stock solutions) for each solid sample using ultrapure water (UP water). The stock solution was diluted at 12.5-fold for the ore-1, 16.6-fold for the ore-2, 25-fold for the ore-3, 6.6-fold for the sediment and 16.6-fold for the electronic waste samples before the analysis because of a high iron and copper ion concentration. The diluted samples were analysed using the same general procedure described in Section 2.4. The determination of Pd(II) and Au(III) ions in the final measurement solution (0.5 mL) was performed by FAAS.
Sea and stream water samples were collected in polytetrafluoroethylene containers from Trabzon, Turkey. Water samples were collected in acid leached polyethylene bottles, and acidified to a pH level with 0.1 mol L−1 HNO3 in order to prevent the adsorption of metal ions on the walls of the flask. The water samples were filtered through a 0.45 mm cellulose nitrate before analyses.
2.6. Preparation of standard reference material
In order to confirm the validity of the developed procedure, the method has been applied to the determination of the content of gold and palladium in the Certified Reference Material (CRM-SA-C sandy soil C). An accurately measured sample (0.4 g) of reference matter was dissolved in a microwave digestion system mixture of HNO3 (3.5 mL), HCl (3.5 mL) and HF (1.0 mL). The solution was evaporated to near dryness and filtrated by adding UP-water. The residue was completed to 25 mL (stock solution) volumetric flask with UP-water. An aliquot of 1.25 mL (for Au) and 8.0 mL (for Pd) of this solution was diluted to a final volume of 10 mL, followed by applying the preconcentration procedure.
3. Results and discussion
The selective sensing of heavy metal ions is a very important topic in various chemical systems. We know that organic compounds, having sulphur and nitrogen donor atoms have the capacity to undergo complexation with soft metal cations.25 Pd2+ and Au3+ ions are especially known as soft metal cations and prefer nitrogen and sulfur donor atoms in complexation with ligands.26 Herein, we report the newly synthesized N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea ligand for the determination of Au3+ and Pd2+ ions combined using DLLME. Furthermore, in order to obtain high extraction efficiency, different parameters like the volume and nature of both the disperser and extraction solvents, as well as the pH of the aqueous solution, the concentration of chelating agent, and the volume of the sample were optimized.
3.1. Characterization of the complexes
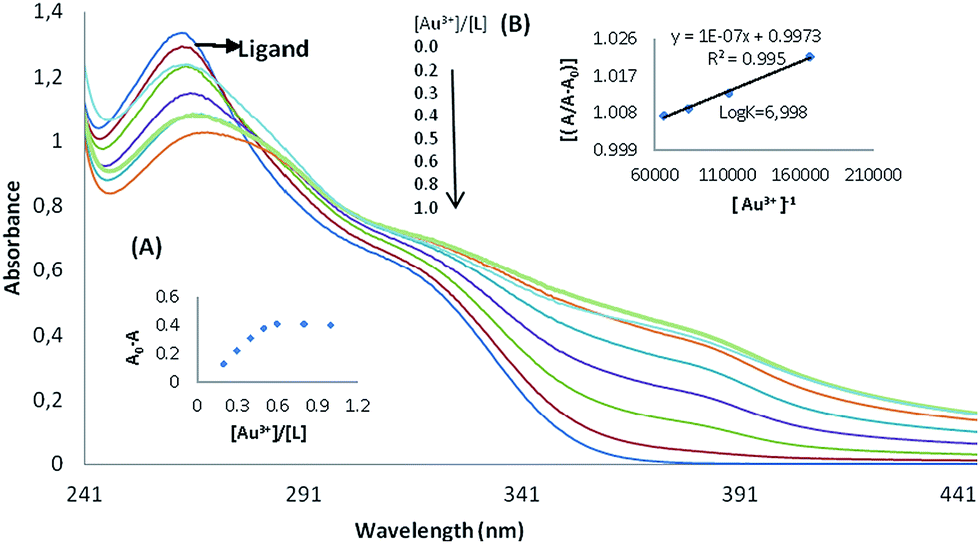
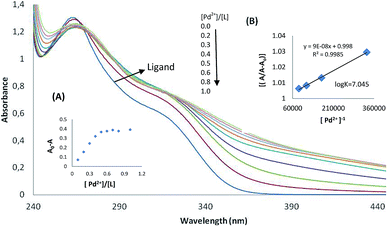
The absorption spectrum of the MPPT in methanol has not displayed absorption signals at 370–440 nm. Fig. 2 and 3 shows the effect of Au3+ and Pd2+ cations on the absorption spectra of the ligand. The presence of 10 equivalents of Au3+ and Pd2+ ions cations caused a decrease in the absorption of the ligand after 370 nm. The stoichiometry of the developed complexes, i.e. Au3+–MPPT and Pd2+–MPPT in methanol–water solutions, were determined by the mole ratio method. The general complex formation equilibrium is assumed by the following eqn (1):| | |
Maqn+ + nMPPTorg ↔ [M(MPPT)n]org
| (1) |
 |
| | Fig. 2 Variation of absorbance of MPPT with concentration of Au3+, added as 0–1 equivalent of HAuCI4, MPPT concentration: 6 × 10−5 mol L−1, measurements were carried out at 380 nm. | |
 |
| | Fig. 3 Variation of absorbance of MPPT with concentration of Pd2+, added as 0–1 equivalent of Pd(NO3)2, MPPT concentration: 6 × 10−5 mol L−1, measurements were carried out at 380 nm. | |
Fig. 2A and 3A (inset) shows the molar-ratio plot for Au3+ and Pd2+, Ao and A are the absorbance of free ligand and the absorbance of the solution involving metal cation, respectively. The inflection point was 0.5 ([Au3+]/[L]) and ([Pd2+]/[L]). It can thus be concluded that ligand formed a stable 1:2 (M:L) complex with Au3+ and Pd2+. In order to determine the complex stability constant according to valuer's method,27 the ratio Ao/(AoA) was plotted versus 1/[M], which yielded a straight line (Fig. 2B and 3B). Ao and A are the absorbance of the free ligand and the absorbance of the solution containing the Au3+ and Pd2+. The stability constant was calculated from the ratio intercept/slope. The result is the stable complex of an Au3+ complex with a logK value of 6.998, and a Pd2+ complex with a logK value of 7.045. All absorbance results were examined at 380 nm.
3.2. Effect of pH
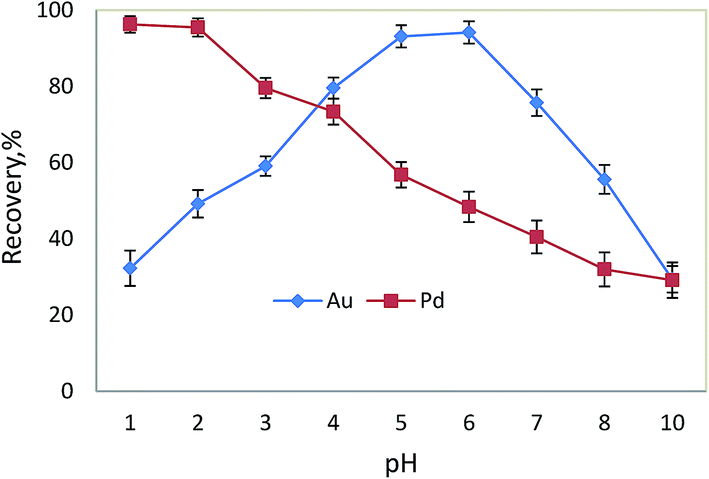
The performance of DLLME process is also tied with the formation of the hydrophobic complex and stability of the complex. The pH of the solution system plays an important role in this step.28 In order to evaluate the effect of the pH on the extraction efficiency, the pH of 10 mL sample solutions containing 0.5 μg gold and palladium ions was adjusted to fit into the range of 1–10. As can be seen in Fig. 4, the red and blue lines have crossed at pH 3–4, but the recoveries were not quantitative for either ion. The maximum recoveries for both ions were obtained in the pH range of 5–6 and 1–2 for Au and Pd, respectively.
 |
| | Fig. 4 Effect of pH of the sample solution on the Au3+ (0.5 μg) and Pd2+ (0.5 μg) signals; sample volume 10 mL, amount and type of extraction solvent: 150 μL of CCl4, amount and type of disperser solvent: 1000 mL of methanol for Au, 1000 mL of ethanol for Pd, amount of MPPT: 125 μL (0.5 mg, 0.4%), centrifugation time and rate: 3 min and 3000 rpm, standing time 5 min, N = 3. | |
3.3. Effect of amount of the MPPT
The amount of complexing agent is a significant parameter that has been found to affect the extraction performance in the preconcentration methods. N-(6-Morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea (MPPT) was used for this purpose. The effects of the MPPT amount on DLLME were studied in the range of 0–175 μL of MPPT (0.4%, w/v). The results were showed in Fig. 5. The recovery values for Au(III) and Pd(II) at the DLLME procedure without MPPT were below 5%. It was found that the recovery of Pd2+ and Au3+ increased with the increasing of the MPPT amount, and then it remained constant at 100 μL of the 0.4% ligand. Therefore, 125 μL of MPTT (0.5 mg) amount was chosen for further experiments.
 |
| | Fig. 5 The effect of the amount of MPPT on the extraction of Au3+ (0.5 μg) and Pd2+ (0.5 μg); sample volume 10 mL, amount and type of extraction solvent: 150 μL of CCl4, amount and type of disperser solvent: 1000 mL of methanol for Au, 1000 mL of ethanol for Pd, centrifugation time and rate: 3 min and 3000 rpm, standing time 5 min, N = 3. | |
3.4. The effect of type and volume of the disperser solvent
The most important point for the selection of a suitable disperser solvent is its mutual miscibility in both extraction solvent and aqueous sample. For this purpose, different solvents such as methanol, ethanol, acetone, acetonitrile and tetra hydro furan were tested. Thus, under the same experimental conditions, a series of sample solutions were prepared by using 1200 μL of each disperser solvent containing 125 μL of carbon tetrachloride. The recommended procedure was then followed. Among the four candidate solvents, methanol and ethanol were selected as disperser solvents for Au and Pd, respectively (see Fig. 6a).
 |
| | Fig. 6 (a) Effect of type of disperser solvent (b) effect of volume of methanol for Au and ethanol for Pd; sample volume 10 mL (0.5 μg Au3+ and 0.5 μg Pd2+), amount of MPPT: 125 μL (0.5 mg), 150 μL of CCl4, N = 3. | |
The effect of the volume of methanol on the extraction efficiency of Au and Pd ions was examined. To obtain the optimized volume of methanol, various experiments were performed using different volumes of methanol (200–3000 μL) containing 150 μL CCl4 for Au. It was found that (Fig. 6b), at a low volume of methanol, the cloudy solution was not formed completely while, at high methanol volume, the solubility of carbon tetrachloride in the aqueous solution was increased. A volume of methanol of 1200 μL was finally chosen. The same results were obtained using the volume of ethanol (200–3000 μL). The optimum ethanol volume of 1200 μL was selected for Pd.
3.5. The effect of type and volume of the extraction solvent
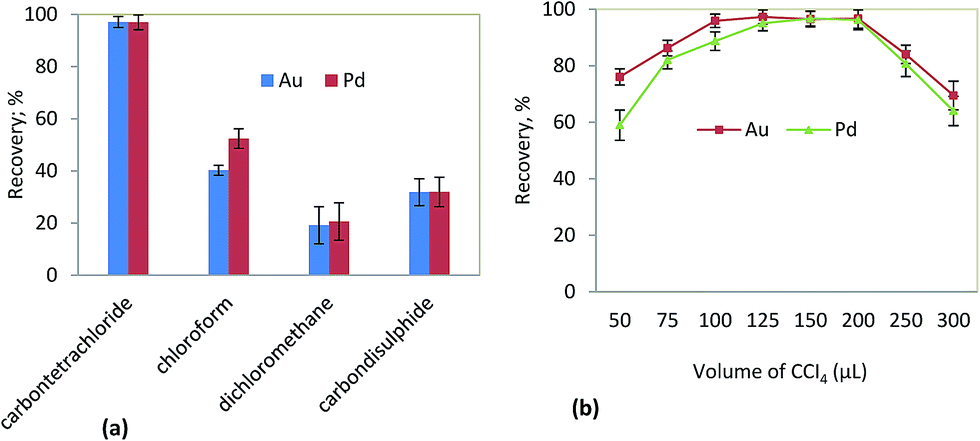
The type of extraction solvent used in DLLME is an essential consideration for efficient extraction. It should have low solubility in water, high affinity to analytes, and a higher density than water. Chloroform (CHCl3), carbon disulfide (CS2), carbontetrachloride (CCl4) and dichloromethane (CH2Cl2) were studied as extraction solvent using 1200 mL of ethanol, and methanol as the dispersive solvent. The results revealed that carbon tetrachloride has the highest extraction efficiency (97.0%) in comparison with carbon disulfide (31.8%), dichloromethane (19.2%), and chloroform (40.2%) in Fig. 7a. Thus, CCl4 was selected for further experiments.
 |
| | Fig. 7 (a) Influence of type of extraction solvent on DLLME (b) influence of volume of CCl4 on the extraction of Au and Pd; sample volume 10 mL, pH = 5.5 for Au3+, pH = 1.5 for Pd2+, amount of MPPT: 125 μL 0.5 mg, N = 3. | |
A suitable volume of extraction solvent is crucial in this method. The effect of carbon tetrachloride volumes on the extraction performance was studied in the range of 50–300 μL. According to Fig. 7b, the recovery of Au and Pd ions increased significantly by increasing the volume of carbon tetrachloride. It was found that the best quantitative recovery was >97% for Au and >95% for Pd when using 125 μL CCI4. As result, in order to obtain a higher extraction recovery value, 125 μL of the extraction solvent was used as the optimal volume in subsequent experiments.
3.6. Effect of the centrifugation rate and time, standing time
Due to the formation of the microdroplets in DLLME, the large surface of the contact between both phases leads to a very fast mass transfer process and generally offers a fast extraction procedure. Therefore, this method is independent from time, one of its most important advantages.29,30 The extraction recovery was investigated by varying the stirring rate in the range of 1000, 4000 rpm. The recoveries were slightly enhanced up to 3000 rpm and remained at a 4000 rate. Finally, a stirring rate of 3000 rpm was chosen. In the next step, the extraction recovery was studied at time intervals of between 1 and 15 min using a stirring rate of 3000 rpm. The results showed that the agitation time had no significant impact of the extraction recovery and, therefore, a practical time of 3 min was selected for further studies.
3.7. Effect of sample volume
To get high preconcentration factor, sample volume is a key factor.31–34 The preconcentration capability of DLLME procedure was further considered by studying the effect of an aqueous volume on the recovery of 0.5 μg of gold and 0.5 μg of palladium from different sample volumes of between 5 mL to 30 mL. The results showed that the extraction was quantitative with the aqueous phase volume up to 10 mL. Based on the final extract volume (0.5 mL) and the maximum sample volume, the extraction was quantitative (10 mL) and therefore a theoretical preconcentration factor of 20 was determined for both ions.
3.8. Interference studies
Matrix effects35–38 are a big problem in the instrumental determination of gold and palladium at trace levels. The efficiency of the development method in preconcentration of gold and palladium in the presence of various cations and anions was examined. A 10 mL model solution, which included 0.5 μg of Au(III) and Pd(II) as well as different amounts of other ions was prepared and the proposed extraction method was applied to the solution. The obtained results are shown in Table 1. As the results show, several species did not interfere even at high concentrations. This is applicable to the analysis of different samples. No interferences were observed from most of the ions tested except for Fe3+, Cu2+, Ni2+ and Mn2+ ions. In order to eliminate the interference effects of these ions, EDTA, NaF were used as the masking agent.39,40
Table 1 Tolerant limits of coexisting ions for the extraction 50 μg L−1 of palladium and gold ions, N = 3
| Ion |
Added as |
Concentration (mg L−1) |
Au3+, recovery (%) |
Pd2+, recovery (%) |
| Masked with 15 mg EDTA. Masked with 20 mg NaF. Ions concentration for Pd. |
| Na+ |
NaCl |
15000 |
100 ± 2 |
99 ± 2 |
| K+ |
KCl |
5000 |
97 ± 3 |
97 ± 2 |
| Ca2+ |
CaCl2 |
500 |
97 ± 3 |
97 ± 3 |
| Mg2+ |
Mg(NO3)2 |
500 |
97 ± 1 |
97 ± 1 |
| Ba2− |
BaCl2 |
50 |
96 ± 2 |
96 ± 2 |
| Mn2+ |
Mn(NO3)2 |
40a–25c |
99 ± 3 |
99 ± 2 |
| Zn2+ |
Zn(NO3)2 |
20–25c |
96 ± 1 |
96 ± 1 |
| Al3+ |
Al(NO3)3 |
25 |
97 ± 3 |
97 ± 3 |
| Pb2+ |
Pb(NO3)2 |
20 |
97 ± 2 |
97 ± 2 |
| Cu2+ |
Cu(NO3)2 |
80a–100c |
101 ± 2 |
93 ± 2 |
| Fe3+ |
Fe(NO3)3 |
100b–100c |
105 ± 2 |
95 ± 3 |
| Ni2+ |
Ni(NO3)2 |
30a–25c |
101 ± 3 |
95 ± 2 |
| EDTA |
Na2H2Y·2H2O |
1500 |
98 ± 3 |
— |
| Cr3+ |
Cr(NO3)3 |
20–25c |
94 ± 2 |
96 ± 2 |
| SO42− |
Na2SO4 |
750–1000c |
94 ± 2 |
96 ± 2 |
| F− |
NaF |
3000 |
102 ± 3 |
— |
| PO43− |
Na3PO4 |
400 |
95 ± 23 |
94 ± 2 |
3.9. Analytical figures of merit
Under optimum conditions, the calibration equation observed by the DLLME procedure were found A = 0.6438C(Au) + 0.0020 (R2 = 0.997) for Au(III) and A = 0.7415C(Pd) + 0.0035 for Pd(II). Where A is absorbance for Au and Pd ions in the enriched phase, C(Au) and C(Pd) are gold and palladium have concentrations of μg L−1 in the sample solution. The linear calibration equation without preconcentration was A = 0.0334C(Au) + 0.0030 (R2 = 0.997) for Au and A = 0.0380C(Pd) + 0.0026 for Pd. The theoretical enrichment factor was calculated by the ratio of volumes of the sample (10.0 mL) and the final solutions (0.5 mL). The experimental enrichment factor was calculated based on the slopes of the calibration curves with or without the extraction for both ions.41 The calibration graph was prepared against the aqueous standards by submitting to the same DLLME procedure. Blank determinations were carried out as parallel to the measurements made for the calibration standards and sample solutions. The blank signals were 0.0024A and 0.0020A for gold and palladium in FAAS. Limits of detection (LODs) were calculated based on 3Sb/m (Sb is the standard deviation of ten replicate blank measurements and m is the slope of the calibration graph) and found as 1.75 for Au and 1.65 μg L−1 for Pd. The relative standard deviation (RSD) for the ten replicate measurements of 10 mL of standard solutions, including 0.5 μg Au(III) and Pd(II) ions was found to be 2.77 and 2.52%, respectively (Table 2).
Table 2 Analytical figures of the DLLME-FAAS method for determination of gold and palladium
| Ion |
Characteristics |
Figures of merit |
| Au3+ |
Linear range with DLLME (μg L−1) |
30–230 |
| Linear range without DLLME (μg mL−1) |
0.4–10 |
| Experimental enrichment factor |
19.3 |
| Theoretical enrichment factor |
20 |
| Limit of detection (LOD, μg L−1), 3Sb/m |
1.75 |
| Limit of quantification (LOD, μg L−1) |
5.76 |
| Precision (RSD, %), 50 μg L−1, n: 10 |
2.77 |
| Sample volume (mL) |
10 |
| Pd2+ |
Linear range with DLLME (μg L−1) |
25–200 |
| Linear range without DLLME (μg mL−1) |
0.3–10 |
| Experimental enrichment factor |
19.5 |
| Theoretical enrichment factor |
20 |
| Limit of detection (LOD, μg L−1), 3Sb/m |
1.65 |
| Limit of quantification (LOD, μg L−1) |
5.43 |
| Precision (RSD, %), 50 μg L−1, n: 10 |
2.52 |
| Sample volume (mL) |
10 |
3.10. Applications of the method
The Certified Reference Material (CRM-SA-C sandy soil C) was analyzed to validate the accuracy of the proposed method. The analytical results are shown in Table 3. The experimental results were in agreement with the values of the CRM 25 μg g−1 for Au3+, 4 μg g−1 for Pd2+ (the values are not certified. They are provided for information purposes only). The presented microextraction method was applied to the determination of palladium and gold in seawater, stream water, ore, sediment, and electronic waste samples. Recovery studies were developed for samples spiked at known concentration levels of Pd and Au. The results are shown in Table 4. The analytical results demonstrated that the suggested method was applied to the analysis of aqueous and solid samples (Table 5).
Table 3 The analysis results of CRM-sandy soil C certified reference material (N: 3)
| Ion |
Certified value (μg g−1) |
Found (μg g−1) |
Recovery (%) |
| Non certified values, provided for information only. Diluted 3.3 fold and added 10 mg NaF because of high iron content. Diluted 1.25 fold. |
| Au |
25a,b |
23.1 ± 1.5 |
92.4 |
| Pd |
4a,b,c |
3.8 ± 0.2 |
95.0 |
Table 4 Addition and recovery tests for the microextraction of Au3+ and Pd2+ in water and solid samples (N: 5)
| Sample |
Added (μg) |
Au |
Pd |
| Found (μg) |
Recovery (%) |
Found (μg) |
Recovery (%) |
| 12.5 fold diluted for Au, 6.6 fold diluted for Pd. 10 mg EDTA, 10 mg NaF added and 16.6 fold diluted for Au, only 6.6 fold diluted for Pd. 10 mg EDTA, 10 mg NaF added and 25 fold diluted for Au, only 6.6 fold diluted for Pd. 10 mg EDTA added and 20 fold diluted for Au, only 16.6 fold diluted for Pd. 10 mg EDTA added and 20 fold diluted, only 6.6 fold diluted for Pd. |
| Stream water-1 |
0 |
<LOD |
— |
<LOD |
— |
| 0.5 |
0.48 ± 0.02 |
96 ± 4 |
0.48 ± 0.01 |
96 ± 2 |
| 1.0 |
0.97 ± 0.02 |
97 ± 2 |
0.98 ± 0.02 |
98 ± 2 |
| Stream water-2 |
0 |
<LOD |
— |
<LOD |
— |
| 0.5 |
0.48 ± 0.01 |
96 ± 2 |
0.48 ± 0.01 |
96 ± 2 |
| 1.0 |
0.97 ± 0.03 |
97 ± 3 |
0.97 ± 0.02 |
98 ± 2 |
| Sea water |
0 |
<LOD |
— |
<LOD |
— |
| 0.5 |
0.49 ± 0.01 |
98 ± 2 |
0.48 ± 0.01 |
96 ± 4 |
| 1.0 |
0.98 ± 0.02 |
98 ± 2 |
0.96 ± 0.02 |
98 ± 2 |
| Ore-1a |
0 |
0.30 ± 0.01 |
— |
<LOD |
— |
| 0.5 |
0.78 ± 0.02 |
96 ± 2 |
0.48 ± 0.02 |
96 ± 4 |
| 1.0 |
1.25 ± 0.04 |
95 ± 2 |
0.98 ± 0.00 |
98 ± 0 |
| Ore-2b |
0 |
0.12 ± 0.01 |
— |
<LOD |
— |
| 0.5 |
0.60 ± 0.02 |
96 ± 4 |
0.49 ± 0.01 |
98 ± 2 |
| 1.0 |
1.10 ± 0.04 |
98 ± 4 |
0.98 ± 0.03 |
98 ± 3 |
| Ore-3c |
0 |
0.49 ± 0.03 |
— |
<LOD |
— |
| 0.5 |
0.96 ± 0.02 |
94 ± 3 |
0.48 ± 0.01 |
96 ± 2 |
| 1.0 |
1.46 ± 0.02 |
97 ± 3 |
0.95 ± 0.02 |
95 ± 2 |
| Electronic wasted |
0 |
0.40 ± 0.02 |
|
0.39 ± 0.01 |
|
| 0.5 |
0.87 ± 0.04 |
94 ± 2 |
0.87 ± 0.02 |
96 ± 4 |
| 1.0 |
1.37 ± 0.05 |
97 ± 2 |
1.35 ± 0.03 |
96 ± 3 |
| Sedimente |
0 |
<LOD |
— |
<LOD |
— |
| 0.5 |
0.47 ± 0.02 |
94 ± 4 |
0.48 ± 0.02 |
96 ± 4 |
| 1.0 |
0.96 ± 0.02 |
96 ± 2 |
0.98 ± 0.02 |
98 ± 2 |
Table 5 The application of the presented method for the analysis of gold(III) and palladium(II) in water and solid samples, N: 3
| Sample |
Concentration, μg g−1, Au |
Concentration, μg g−1, Pd |
| Stream water-1 |
<LOD |
<LOD |
| Stream water-1 |
<LOD |
<LOD |
| Sea water |
<LOD |
<LOD |
| Ore-1 |
23.1 ± 1.1 |
<LOD |
| Ore-2 |
12.8 ± 0.8 |
<LOD |
| Ore-3 |
76.3 ± 4.2 |
<LOD |
| Electronic waste |
132.2 ± 5.2 |
108.6 ± 4.1 |
| Sediment |
<LOD |
<LOD |
3.11. Comparison with other preconcentration techniques
The presented DLLME method with other sample preparation techniques, in combination with FAAS for the extraction and determination of gold and palladium, was compared in Table 6. As can be seen from Table 6, they are mostly one-element methods, with the exception of our presented method. The DLLME-FAAS has a comparatively low detection limit (1.65 μg L−1 for Pd and 1.75 μg L−1 for Au) except ref. 36 and 40. The lower detection limit was reported with some references that could be the result of the determination system. We have good linear dynamic ranges and a good enrichment factor for both ions. The previously reported methods only were used in water samples for the preconcentration of Au or Pd ions. The proposed DLLME method combined with FAAS can be used for several environmental liquid and solid samples. All these results demonstrate that the DLLME is indeed simple, quick, efficient, easy to use, and environmentally friendly – involving minimum usage of organic solvents. Also, this methodology is a reproducible, simple, and low cost technique for laboratories doing routine analysis of gold and palladium in various sample types such as, water, ore, sediment and electronic waste.
Table 6 Comparison of proposed DLLME with published preconcentration methodsa
| Ion |
Analytical technique/detection method |
EF |
LOD, μg L−1 |
LDR, μg L−1 |
RSD, % |
Ref. |
| LDR: linear dynamic range, LOD: limit of detection, IP-DLLME: ion-pair dispersive liquid–liquid microextraction, UIL-DLLME: ionic liquid-based dispersive liquid–liquid microextraction. |
| Pd2+ |
DLLME/FAAS |
45.7 |
90 |
100–2000 |
0.7 |
42 |
| Pd2+ |
DLLME/FAAS |
138 |
19 |
40–800 |
3.2 |
43 |
| Pd2+ |
DLLME/GFAAS |
166.5 |
0.02 |
0.05–1.0 |
3.5 |
44 |
| Au3+ |
IL-LME/FAAS |
10 |
72 |
190–3820 |
— |
45 |
| Au3+ |
IL-DLLME/FAAS |
23.7 |
0.13 |
0.9–400 |
— |
46 |
| Au3+ |
DLLME/GFAAS |
48.7 |
0.0048 |
0.02–40 |
4.1 |
47 |
| Au3+ |
IP-DLLME/FAAS |
40 |
1.8 |
8–100 |
3.2 |
48 |
| Pd2+ |
DLLME/FAAS |
20 |
1.65 |
30–230 |
2.52 |
This work |
| Au3+ |
DLLME/FAAS |
20 |
1.75 |
25–200 |
2.77 |
This work |
4. Conclusion
In this procedure, the N-(6-morpholin-4-ylpyridin-3-yl)-N′-phenylthiourea (MPPT) reagent was successfully used as a complexing agent for the preconcentration of gold(III) and palladium(II) ions. The DAT reagent was synthesized first in our laboratory. Complexation behavior of MPPT towards the selected series of metal ions worked using the A novel dispersive liquid–liquid microextraction procedure. Experimental observations revealed that it is efficient and selective for Pd2+ and Au3+ ions in aqueous solutions. It has fast kinetic properties and is also stable in solutions for long periods of time. The DLLME approach associated with FAAS detection can be proposed as an extraction technique tool for the determination of trace elements with a short extraction time, good precision, minimal organic solvent use, and environmentally friendliness factor. This is a novel method suitable for the simple and accurate determination of gold and palladium in a variety of water and solid samples.
Acknowledgements
Financial support of the Unit of the Scientific Research Projects of Karadeniz Technical University (Project no: 1223) is gratefully acknowledged.
References
- H. Fazelirad and M. Ali Taher, Talanta, 2013, 103, 375–383 CrossRef CAS PubMed.
- K. Pyrzynska, Anal. Chim. Acta, 2012, 741, 9–14 CrossRef CAS PubMed.
- P. Liang, E. Zhao, Q. Ding and D. Du, Spectrochim. Acta, Part B, 2008, 63, 714–717 CrossRef.
- C. R. M. Rao and G. S. Reddi, Trends Anal. Chem., 2000, 19, 565–586 CrossRef CAS.
- K. Ravindra, L. Bencs and V. Grieken, Sci. Total Environ., 2004, 318, 1–43 CrossRef CAS PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press, Oxford, New York, 1989 Search PubMed.
- J. D. Lee, Concise Inorganic Chemistry, ELBS with Chapman and Hall, London, 5th edn, 1996 Search PubMed.
- M. B. Gómez, M. M. Gómez and M. A. Palacios, Anal. Chim. Acta, 2000, 404, 285–294 CrossRef.
- P. Liang and E. Zhao, Microchim. Acta, 2011, 174, 153–158 CrossRef CAS.
- M. Ezoddin, B. Majidi and K. Abdi, J. Mol. Liq., 2015, 209, 515–519 CrossRef CAS.
- N. Ozturk, V. N. Bulut, C. Duran and M. Soylak, Desalination, 2011, 270, 130–134 CrossRef CAS.
- A. N. Anthemidis, D. G. Themelis and J. A Stratis, Talanta, 2001, 54, 37–43 CrossRef CAS PubMed.
- M. S. El-Shahawi, A. S. Bashammakh and S. O. Bahaffi, Talanta, 2007, 72, 1494–1499 CrossRef CAS PubMed.
- L. Elci, M. Soylak and E. B. Buyuksekerci, Anal. Sci., 2003, 19, 1621–1624 CrossRef CAS PubMed.
- H. B. Senturk, A. Gundogdu, V. N. Bulut, C. Duran, M. Soylak, L. Elçi and M. Tufekci, J. Hazard. Mater., 2007, 149, 317–323 CrossRef CAS PubMed.
- L. Tavakoli, Y. Yamini, H. Ebrahimzadeh, A. Nezhadali, S. Shariati and F. Nourmohammadian, J. Hazard. Mater., 2008, 152, 737–743 CrossRef CAS PubMed.
- C. Zeng and L. Tang, Anal. Lett., 2013, 46, 1442–1453 CrossRef CAS.
- M. Bagheri, M. H. Mashhadizadeh and S. Razee, Talanta, 2003, 60, 839–844 CrossRef CAS PubMed.
- M. Ghaedi, M. Ghayedi, S. N. Kokhdan, R. Sahraei and A. Daneshfar, J. Ind. Eng. Chem., 2013, 19, 1209–1217 CrossRef CAS.
- S. Fragueiro, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2004, 59, 851–855 CrossRef.
- M. Rezaee, Y. Assadi, M. R. M. Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- H. Eskandari, Turk. J. Chem., 2012, 36, 631–643 CAS.
- P. Liang, E. Zhao and F. Li, Talanta, 2009, 77, 1854–1857 CrossRef CAS PubMed.
- S. Kagaya, D. Takata, T. Yoshimori, T. Kanbara and K. Tohda, Talanta, 2010, 80, 1364–1370 CrossRef CAS PubMed.
- A. Ohki, M. Takagi and K. Ueno, Anal. Chim. Acta, 1984, 159, 245–251 CrossRef CAS.
- C. Ozdemir, Ş. Saçmacı, Ş. Kartal and M. Saçmacı, J. Ind. Eng. Chem., 2014, 20, 4059–4065 CrossRef CAS.
- J. Bourson and B. Valeur, J. Phys. Chem., 1989, 93, 3871–3875 CrossRef CAS.
- M. D. Farahani, F. Shemirani and M. Gharehbaghi, Talanta, 2013, 109, 121–127 CrossRef PubMed.
- M. Saraji and M. K. Boroujeni, Anal. Bioanal. Chem., 2014, 406, 2027–2066 CrossRef CAS PubMed.
- B. Majidi and F. Shemirani, Talanta, 2012, 93, 245–251 CrossRef CAS PubMed.
- M. Soylak, E. B. Buyuksekerci and L. Elci, Asian J. Chem., 2004, 16, 1625–1629 CAS.
- S. H. Ahmadi, A. M. Haji Shabani, S. Dadfarnia and M. Taei, Turk. J. Chem., 2007, 31, 191–199 CAS.
- M. Soylak, L. Elci, I. Narin and M. Dogan, Asian J. Chem., 2001, 13, 699–703 CAS.
- Y. Yamini, M. Moradi and E. Tahmasebi, Anal. Chim. Acta, 2012, 728, 26–30 CrossRef CAS PubMed.
- M. Soylak and M. Tuzen, J. Hazard. Mater., 2008, 152, 656–661 CrossRef CAS PubMed.
- M. Soylak and L. Elci, Int. J. Environ. Anal. Chem., 1997, 66, 51–59 CrossRef CAS.
- E. Kazemi, S. Dadfarnia and A. M. Haji Shabani, Talanta, 2015, 141, 273–278 CrossRef CAS PubMed.
- L. Elci, D. Sahan, A. Basaran and M. Soylak, Environ. Monit. Assess., 2007, 132, 331–338 CrossRef CAS PubMed.
- E. Yilmaz and M. Soylak, RSC Adv., 2014, 4, 47396–47401 RSC.
- G. S. Kamble, S. S. Kolekar, S. H. Han and M. A. Anuse, Talanta, 2010, 81, 1088–1095 CrossRef CAS PubMed.
- N. Shokoufi, F. Shemirani and Y. Assadi, Anal. Chim. Acta, 2007, 597, 349–356 CrossRef CAS PubMed.
- T. A. Kokya and K. Farhadi, J. Hazard. Mater., 2009, 169, 726–733 CrossRef CAS PubMed.
- K. Gholamreza and S. Kaveh, J. Inclusion Phenom. Macrocyclic Chem., 2014, 79, 185–191 CrossRef.
- R. J. Mohammad, A. Yaghoub and R. K. Reyhaneh, J. Chem., 2013, 671743 Search PubMed.
- O. Sha and X. Zhu, Anal. Lett., 2014, 47, 1052–1062 CrossRef CAS.
- O. Sha, J. Chen, L. Chen and S. Li, J. Iran. Chem. Soc., 2015, 12, 1391–1398 CrossRef CAS.
- A. Hamid and A. T. Mohammad, Microchem. J., 2012, 103, 185–190 CrossRef.
- A. Hol, A. A. Kartal, A. Akdogan, A. Elçi, T. Arslan and L. Elci, Acta Chim. Slov., 2015, 62, 196–203 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.