
CO2 capture under humid conditions in NH2-MIL-53(Al): the influence of the amine functional group†
Antonio Zárate‡
a,
Ricardo A. Peralta‡a,
Peter A. Baylissb,
Rowena Howieb,
Mayra Sánchez-Serratosa,
Paulina Carmona-Monroya,
Diego Solis-Ibarra*a,
Eduardo González-Zamora*c and
Ilich A. Ibarra*a
aInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, México D. F., Mexico. E-mail: argel@unam.mx
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK
cDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, Iztapalapa, C. P. 09340, México D. F., Mexico
First published on 19th January 2016
Abstract
The hydrostable MIL-53(Al) and NH2-MIL-53(Al) CO2 capture properties were confirmed by kinetic uptake experiments, under different humidity conditions with maximum CO2 captures of approximately 6.0 wt% and 4.6 wt%, respectively, at 5% RH at 30 °C. In the case of MIL-53(Al) this corresponds to a 1.7-fold increase (CO2 capture) in comparison to anhydrous conditions. NH2-MIL-53(Al) exhibited a considerably stronger affinity to water than MIL-53(Al), and its ability to capture CO2, under humid conditions, was significantly reduced. The in situ FTIR experiments show how the hydrophobicity of the pores within MIL-53(Al) enhanced and sustained the CO2 adsorption capabilities of the material under more realistic CO2 capture conditions.
Introduction
One of the main contributors to climate change (global warming) is carbon dioxide (CO2) emission from fossil fuel combustion.1a Indeed, fossil fuel combustion provides more than 85% of the energy required for industrial applications.1b The CO2 levels have increased, from 1990 to date, up to 45% (ref. 2) as a result of human activities e.g. steel production and coal-fired power plants.1 Thus, the rigorous reduction of these CO2 emissions is fundamental in order to minimise the risk that global warming represents to our society. One strategy towards reducing CO2 emissions is to capture and permanently sequester CO2. To achieve this goal, the development of new methodologies for efficient CO2 capture has been addressed by many international initiatives.3 Poliakoff4 proposed ‘the twelve principles of CO2 chemistry’ where CO2 capture represents one of these principles (maximise integration) and thus, highly porous solid materials are very promising candidates for the solution of this worldwide task.Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) have received significant attention as potentially valuable CO2 capture media,5 since these materials can be tailored as a function of the size, shape and chemical composition of the pores.6 One of the main limitations that PCPs show, when used as CO2 capture materials, is their water instability.7,8 For example H2O molecules could block the binding adsorption sites and as a result of it, these obstruct the adsorption of the desired target-molecules.8 Additionally, water could reallocate the bound ligand, leading to the disintegration of the PCP structure.8 Since moisture is always present in the environment, it must be considered for any adsorption and storage process. Therefore, water stability of PCPs is one key property for any CO2 capture applications. As an example, industrial flue gas which results from the burning of fossil fuels, is typically saturated with 5–7% of H2O.9
Recently, there is a substantial amount of PCPs that have shown relatively good stability to water: NOTT-401,10 MIL-100,11 UiO-66,12 InOF-1,13 MIL-101,14 MIL-53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and Cu(bcppm)H2O.16 Thus, some of these moisture-stable PCPs have been used in storage (H2O) technologies for arid environments,17 proton conductivity18 and heat-pumps chillers.19
15 and Cu(bcppm)H2O.16 Thus, some of these moisture-stable PCPs have been used in storage (H2O) technologies for arid environments,17 proton conductivity18 and heat-pumps chillers.19
The combined adsorption of CO2 and water (CO2 + H2O) in PCPs has recently been investigated.20 Commonly, the adsorption of water diminishes the CO2 capture (adsorption). However, Walton,21 LeVan22 and Matzger23 showed that when water adsorption in PCPs is controlled, this can increase the CO2 capture in PCPs. Llewellyn24 studied the CO2 capture (under different relative humidity conditions) in the mesoporous material entitled MIL-100(Fe), and an outstanding 5-fold enhance in the CO2 uptake was achieved. In a comprehensive study about the effect of the functional group on the adsorption behaviour, Walton25 exhibited how the amine functional group acts as a directing agent for the H2O molecules within the pores of the UiO-66 series, which provided more efficient packing. Moreover, Yaghi et al.17 suggested that the existence of hydroxyl functional groups, inside the porous material, increases the affinity of PCPs for H2O.
Férey26 reported for the very first time the MIL-53 porous coordination polymer series. Particularly, the Al(III) based version of MIL-53, entitled MIL-53(Al), is water stable15 and in a previous work,27 we reported the enhanced CO2 capture properties in the presence of water in MIL-53(Al). Thus, in here we selected the amine functionalised MIL-53(Al), NH2-MIL-53(Al), to study the CO2 sequestration under humid conditions and we made a comparison with the non-functionalised material, MIL-53(Al). NH2-MIL-53(Al), first reported by Blom et al.,28 is constructed by infinite trans chains of corner-sharing (via OH groups, μ2-OH) AlO4(OH)2 octahedra interconnected by NH2-BDC2− ligands (NH2-BDCH2 = 2-amino-terephthalic acid, Fig. 1). Thus, three dimensional microporous framework structure is formed with diamond-shaped one dimensional channels (Fig. 1). The material NH2-MIL-53(Al) has previously exhibited very interesting CO2 adsorption properties28,29 and it also has provided promising results in research fields such as heterogeneous catalysis,30 nonlinear optics31 and negative compressibility.32
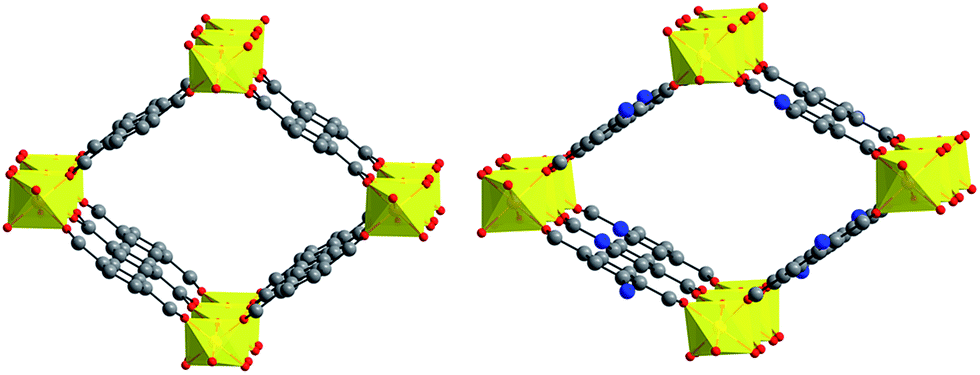 | ||
| Fig. 1 Crystal structures of ht MIL-53(Al) and ht NH2-MIL-53(Al). Aluminium: yellow; oxygen: red; carbon: grey; nitrogen: blue. Hydrogen atoms omitted for clarity. | ||
Experimental section
We previously reported a continuous flow methodology for the synthesis of PCPs in pure water.33 Thus, MIL-53(Al) and NH2-MIL-53(Al) were synthesised using this approach and calcined (extraction of terephthalic and 2-amino-terephthalic acid from the pores) in an oven at 330 °C for 3 days. These samples were labelled as post-synthesised. Thermogravimetric analyses (see Fig. S1, ESI†) and bulk powder X-ray diffraction (PXRD) patterns (see Fig. S2, ESI†) of the post-synthesised MIL-53(Al) and NH2-MIL-53(Al) samples, corroborated that the framework structures of these materials were preserved upon the removal of terephthalic and 2-amino-terephthalic acid. It is worth to mention that, as previously reported,26b the post-synthesised samples of MIL-53(Al) and NH2-MIL-53(Al) correspond to the lt form (room temperature in which water is located within the channels).26b N2 adsorption isotherms for activated MIL-53(Al) and NH2-MIL-53(Al), corresponding to the calcined form (ht)26b empty channels,26b (180 °C under vacuum for 12 h) at 77 K were employed to calculate the BET surface area (0.01 < P/P0 < 0.04) of 1096 m2 g−1 and 780 m2 g−1, respectively. In the case of MIL-53(Al), the BET surface area of 1096 m2 g−1 is consistent with previously reported values that range between 1270 m2 g−1 and 933 m2 g−1.26,33 For sample NH2-MIL-53(Al), the surface area (BET) calculated as 780 m2 g−1, is lower to the value reported by Blom et al.28 of 960 m2 g−1 and higher than the value reported by Gascon and co-workers30 of 675 m2 g−1. Guo et al.29f showed how the synthesis conditions (modifying the ratio of water in the DMF–water mixed solvent system) have a considerable impact on the crystal size and morphology of the material NH2-MIL-53(Al) which is directly observed on the BET surface area from values of 950 m2 g−1 to 1882 m2 g−1.29f Thus, although our BET surface value (780 m2 g−1) is in the lower end of the values reported in the literature, the rapid synthesis of NH2-MIL-53(Al) in only water33 represents a greener methodology with an excellent potential for scale-up.Kinetic uptake experiments were performed using a thermobalance (Q500 HR, from TA) at room temperature (30 °C) with a constant CO2 flow (60 mL min−1). Post-synthesised samples of MIL-53(Al) and NH2-MIL-53(Al) were placed inside the thermobalance and activated by heating from room temperature to 180 °C for 1 h and under a flow of N2 gas (calcined form, ht).26b After the activated sample was cooled down, the temperature was set to 30 °C and a constant CO2 flow (60 mL min−1) was carried out. With a humidity-controlled thermobalance (Q5000 SA, from TA) kinetic uptake experiments at 30 °C with a constant CO2 flow (60 mL min−1) were performed on activated samples (180 °C for 1 h and under a flow of N2 gas) of MIL-53(Al) and NH2-MIL-53(Al).
Ambient conditions (20 °C and 50% RH) Fourier transform Infrared (FTIR) spectroscopy spectra were obtained on a Bruker Alpha spectrometer equipped with an attenuated total reflectance (ATR) accessory at room temperature in the range 500–4000 cm−1. For these experiments activated samples of MIL-53(Al) and NH2-MIL-53(Al), (calcined form, ht)26b were placed on the spectrometer and their behaviour under ambient conditions was monitored by consecutive FTIR measurements (every five minutes) until no change was observed.
Results and discussion
First, dynamic and isothermal (30 °C) CO2 experiments were performed on MIL-53(Al) and NH2-MIL-53(Al) (see Experimental). Fig. 2 shows the kinetic uptake experiments at 30 °C, where weight gain, represents the amount of CO2 captured in both materials. Then, the maximum CO2 uptake for MIL-53(Al) and NH2-MIL-53(Al) were found to be 3.5 wt% and 4.9 wt%, respectively, which were reached after only 10 min and remained constant until 120 min, when the experiment was finished. The amine functionalised NH2-MIL-53(Al) has previously shown to enhance the CO2 uptake over the non-functionalised material.28 Interestingly, Gascon and co-workers34 demonstrated that this CO2 enhancement is not due to the direct interaction of the amine functional (–NH2) group with adsorbed CO2; instead, the presence of the amine modulates the ‘breathing’ behaviour of NH2-MIL-53(Al), which is, the flexibility of the framework. Thus, the amine favours the narrow-pore structure conformation in which, the interactions of the specific adsorbates (e.g. CO2) with the pore walls are higher than for the parent MIL-53(Al), affording higher CO2 uptake.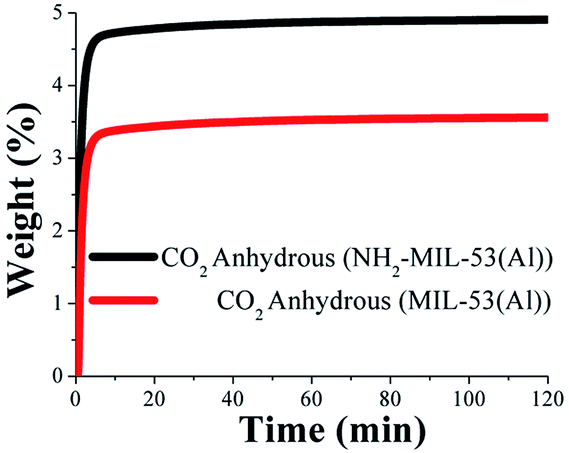 | ||
| Fig. 2 Kinetic uptake experiments performed at 30 °C with a CO2 flow of 60 mL min−1. | ||
Kinetic isotherm experiments at 30 °C and different relative humidities (5, 10 and 30% RH) were carried out. We chose these RH values based upon the investigation of water confined in the isostructural material MIL-53(Cr), proposed by Paesani.35a Through computational infrared spectroscopy, they showed35a that H2O molecules (at low water loadings) interact strongly with the pore walls of this material, MIL-53(Cr), via hydrogen bonding between the μ2-OH functional group and H2O, whereas intermolecular interactions between H2O molecules become considerably stronger at higher loading. Haigis35b postulated by molecular dynamics (MD) that water molecules can form hydrogen bonds with the bridging hydroxo functional groups (μ2-OH) depending on the water loading in the material MIL-53(Cr). In addition, Maurin et al.36 demonstrated by GCMC simulations, in the same material MIL-53(Cr), that at low H2O loadings, the water molecules are homogeneously distributed inside all the pores of the material. Our hypothesis is that at low water loadings, the channels within the materials MIL-53(Al) and NH2-MIL-53(Al) supply a template (with μ2-OH and μ2-OH + –NH2 functional groups, respectively) for a more efficiently packing of H2O molecules and thus, these H2O molecules can then hydrogen-bond to the CO2 molecules enhancing the total CO2 capture.
Then, an activated MIL-53(Al) sample, ht form,26b (180 °C for 1 h and under a flow of N2 gas) was stabilised at 30 °C and 5% RH. After the equilibrium was reached a constant CO2 flow (60 mL min−1) was carried out (see Fig. 3, left). The continuous weight gain (only H2O) starts at 0 min and stabilised at around 25 min. Differently, under anhydrous conditions the CO2 uptake quickly reached stability (10 min, see Fig. 3). Since the diffusion coefficient of water is smaller than CO2, the vapour adsorption (water) process takes considerably more time to reach stability than the gas adsorption process in microporous materials.37 Then, from 25 min to 165 min the H2O uptake (0.5 wt%), which is in good agreement with water adsorption isotherms,38 was invariable (plateau). Next, at 165 min the CO2 flow (60 mL min−1) was opened and a quick weight increase was observed and reached stability at approximately 200 min (see Fig. 3, left). As we previously observed,39 the adsorbed amount of H2O is unchanged after the dosing of H2O + CO2 vapour-gas mixture. Then, from 200 min to the end of the experiment (350 min), the maximum amount of CO2 captured (taking into consideration the water uptake of 0.5 wt%) corresponded to 6.0 wt%. Thus, the CO2 capture was approximately 1.7-fold increased when applying 5% RH (from 3.5 wt% to 6.0 wt%) in comparison to anhydrous conditions. This CO2 capture enhancement, in the presence of RH, can be attributed to CO2 confinement effects induced by H2O molecules.40
 | ||
| Fig. 3 Kinetic uptake experiments carried out at 30 °C and 5% RH for (left) MIL-53(Al), with H2O (blue line) and H2O + CO2 (red line); (right) NH2-MIL-53(Al), with H2O (green line) and H2O + CO2 (red line). | ||
On an activated sample (ht form)26b of NH2-MIL-53(Al), vide infra, kinetic CO2 uptake experiments were carried out at 30 °C and 5% RH. In Fig. 3 (right) the kinetic uptake experiment is shown for NH2-MIL-53(Al) where the constant water weight increase begins at 0 min and stabilises at approximately 50 min. From 50 to 165 min the water uptake was constant and equal to 1.6 wt% (in good agreement with the water adsorption isotherms38). Next, the CO2 flow (60 mL min−1) was started and a sharp weight uptake was observed (see Fig. 3 right). The stabilisation was achieved at around 200 min with a total CO2 capture of 4.6 wt%. This capture, under relative humidity conditions, was certainly lower than under anhydrous conditions (4.9 wt%) representing a 0.3 wt% decrease.
From these kinetic uptake experiments (at 5% RH) on samples MIL-53(Al) and NH2-MIL-53(Al) it is clear that the material with the amine functional group (NH2-MIL-53(Al)) adsorbs more water than the non-functionalised (MIL-53(Al)), 1.6 and 0.5 wt%, respectively, suggesting that the affinity for water of the material NH2-MIL-53(Al) is considerably higher than for MIL-53(Al). In order to confirm this experimental evidence, we decided to run more kinetic CO2 uptake isotherm experiments on MIL-53(Al) and NH2-MIL-53(Al). First, at 30 °C and 10% RH, MIL-53(Al) and NH2-MIL-53(Al), Fig. 4, showed water uptakes (plateau zone of the isotherm) of 1.0 wt% and 3.3 wt%, respectively and in good agreement with water adsorption isotherms,38 with stabilisation times of 50 min and 100 min, respectively (see Fig. 4). After the CO2 flow was switched on, total CO2 uptakes were 5.4 wt% for MIL-53(Al) and 3.5 wt% for NH2-MIL-53(Al), with a stabilisation time of 200 min for both isotherms (Fig. 4).
 | ||
| Fig. 4 Kinetic uptake experiments carried out at 30 °C and 10% RH for (left) MIL-53(Al), with H2O (blue line) and H2O + CO2 (red line); (right) NH2-MIL-53(Al), with H2O (green line) and H2O + CO2 (red line). | ||
Finally, kinetic CO2 uptake isotherm experiments on MIL-53(Al) and NH2-MIL-53(Al) at 30 °C and 30% RH were performed (Fig. 5). Then, the water uptakes for MIL-53(Al) and NH2-MIL-53(Al) were found to be 2.1 wt% and 7.0 wt%, respectively, consistent with water adsorption isotherms.38 In the case of sample MIL-53(Al) the stabilisation time is approximately 100 min (Fig. 5, left) and for sample NH2-MIL-53(Al) there is not a clear stabilisation time since the isotherm did not show a plateau (see Fig. 5, right). After the CO2 was started, the total CO2 uptakes were 4.6 wt% for MIL-53(Al) and 0.5 wt% for NH2-MIL-53(Al). The stabilisation times for MIL-53(Al) were 200 min and for NH2-MIL-53 of approximately 220 min.
 | ||
| Fig. 5 Kinetic uptake experiments carried out at 30 °C and 30% RH for (left) MIL-53(Al), with H2O (blue line) and H2O + CO2 (red line); (right) NH2-MIL-53(Al), with H2O (green line) and H2O + CO2 (red line). | ||
While increasing the relative humidity on the kinetic uptake experiments, in both MIL-53(Al) and NH2-MIL-53(Al) materials, their ability to capture CO2 was undoubtedly reduced. In the case of MIL-53(Al), as we previously reported,27 the presence of water within the micropores of the material enhances the CO2 capture, and in the present work, the optimal relative humidity was found to be 5% RH (6.0 wt% CO2 captured). In contrast, the presence of water in NH2-MIL-53(Al) did not favour the CO2 capture even at low amounts (5% RH). From the dynamic and isothermal experiments it is possible to conclude that the material NH2-MIL-53(Al) exhibits a considerable stronger affinity to water than the non-functionalised material (MIL-53(Al)) and as a consequence of it, at 30 RH% the material was practically saturated with water leaving no room to the CO2 molecules, resulting in a very reduced ability to capture CO2 (only 0.5 wt%) in contrast to anhydrous conditions (4.9 wt%). In addition, due to the preference for the contracted structure (narrow-pore) that the amine functional group (–NH2) enforces34 to the material NH2-MIL-53(Al), the accessibility to the pores is lower than in the non-functionalised material (MIL-53(Al)) which results in lower H2O + CO2 captures at different RH.
To confirm that there was no sample degradation, PXRD measurements and N2 adsorption isotherms (BET surface area) were carried out on all the samples (MIL-53(Al) and NH2-MIL-53(Al)) after CO2 capture experiments (see Fig. S3 and S4, ESI†), which demonstrated that the crystallinity and the surface area of the samples were retained. The PXRD measurements were carried on samples at room temperature, meaning the lt form.26b Since it is necessary to activate (see above) the samples for the BET surface area determination, the form was calcined or ht.26b
We then hypothesised that the different behaviour of these materials in the presence and absence of water can be explained in terms of the hydrophobicity of the pore surface in the PCPs and not in terms of the surface area, pore size or CO2 adsorption under anhydrous conditions. Specifically, the polar NH2 functionality in NH2-MIL-53(Al) makes the pore surfaces hydrophilic in nature, facilitating water diffusion across the pores of the material and providing additional binding sites for the incoming water, effectively blocking the pores and precluding CO2 capture. On the other hand, the more hydrophobic nature of the pores and the scarcity of water binding-sites in the non-functionalised MIL-53(Al) hinder water diffusion therefore limiting water adsorption and leaving room for CO2.
To further investigate the kinetic behaviour of the adsorption process and prove our hypothesis we performed in situ Fourier transform infrared (FTIR) spectroscopy under ambient conditions (20 °C and 50% RH). The experiments were performed on activated samples (ht form)26b that were open to the atmosphere and immediately placed on the spectrometer to collect the first spectra (T = 0, Fig. 6). Then a spectrum was collected every five minutes until no further change was observed. Under these conditions, the samples were simultaneously exposed to ambient humidity, nitrogen, oxygen and small amounts of other gases, among which is CO2 (approximately 400 ppm). The FTIR spectra of the non-functionalised MIL-53(Al) showed minor changes in the O–H region (3000–3600 cm−1), meaning that little to no water was adsorbed under these conditions. Conversely, there is an increase in the CO2 stretching band (2358 cm−1), which clearly indicates that CO2 is the preferred adsorbate over water and therefore, under these conditions, MIL-53(Al) does not immediately transition into its lt (hydrated) form, and it instead adsorbs CO2. On the other hand, the FTIR spectra of NH2-MIL-53(Al) showed no evidence for CO2 absorption, however there is a shift in the frameworks amino N–H and μ2-hydroxo O–H stretches along with a general broadening and signal increase of the overall 3000–3600 cm−1 region. These changes can be attributed to the interaction of the NH2 and OH groups in the pores with the adsorbed water and are consistent with a rapid phase transition from the ht to lt form of NH2-MIL-53(Al).29d Therefore, and in good agreement with our kinetic uptake experiments, NH2-MIL-53(Al) rapidly adsorbs water effectively saturating the material and blocking its pores for CO2 adsorption. Conversely, water adsorption by MIL-53(Al) is less favoured thus, allowing CO2 to be adsorbed on the material. These results are in sharp contrast to the behaviour of these materials under anhydrous conditions, where NH2-MIL-53(Al) adsorbs more CO2 than its non-functionalised counterpart MIL-53(Al).29
 | ||
| Fig. 6 FTIR spectra of activated samples of (left) MIL-53(Al), and (right) NH2-MIL-53(Al), under atmospheric conditions (20 °C and 50% RH). The green lines shows the T = 0 spectra, blue is at T = 10 min and the black one shows the spectra at T = 30 min (saturated). | ||
These results highlight how the hydrophobicity of the pores within a material can have dramatic effects on its overall CO2 adsorption under humid conditions. Specifically, they show how an hydrophobic pore might help enhance and sustain CO2 capture capabilities of a material under more realistic conditions.
Conclusions
The hydrostable Al(III) coordination polymers MIL-53(Al) and NH2-MIL-53(Al) exhibited, by kinetic isotherm CO2 experiments, a total CO2 uptake of 3.5 wt% and 4.9 wt%, respectively, at 30 °C. The CO2 capture properties were evaluated on both materials, MIL-53(Al) and NH2-MIL-53(Al), under different relative humidity conditions (5, 10 and 30% RH) at 30 °C, showing maximum CO2 captures of approximately 6.0 wt% and 4.6 wt%, respectively, at 5% RH. In the case of MIL-53(Al) this CO2 capture under humid conditions corresponds to a 1.7-fold increase in comparison to anhydrous conditions. This CO2 capture enhancement is attributed to CO2 confinement effects induced by H2O40 which occur within the micropores of MIL-53(Al) and in combination with the directing effect of the hydroxo functional groups (μ2-OH) permit CO2 to be accommodated more efficiently.25 Conversely, for sample NH2-MIL-53(Al) the capture of CO2 under relative humidity conditions afforded a decrease of approximately 0.3 wt% when compared to anhydrous conditions. Since NH2-MIL-53(Al) showed a considerable stronger affinity to water than the non-functionalised material, its ability to capture CO2 under humid conditions is significantly reduced and at 30 RH% the material was essentially saturated with water leaving no room to CO2 molecules.The kinetic uptake experiments in combination with in situ FTIR experiments show how the hydrophobicity of the pores within a material can have dramatic effects on its overall CO2 adsorption under humid conditions, specifically, they show how an hydrophobic pore might help enhance and sustain CO2 adsorption capabilities of a material under more realistic conditions.
Acknowledgements
The authors thank Dr A. Tejeda-Cruz (X-ray; IIM-UNAM), CONACyT Mexico (212318), PAPIIT UNAM Mexico (IN100415) for financial support. E. G-Z. thanks CONACyT (236879), Mexico for financial support. Thanks to U. Winnberg (ITAM and ITESM) for scientific discussions. We gratefully acknowledge the receipt of a University of Nottingham 2012 EPSRC Doctoral Prize to P. A. B. and R. A. H. for EPSRC funding. We thank Prof. M. Schröder and Prof. M. Poliakoff for their encouragement.Notes and references
-
(a) S. Chu, Science, 2009, 325, 1599 CrossRef CAS PubMed
; (b) R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS PubMed
- J. Johnson, Chem. Eng. News, 2012, 90, 8 Search PubMed
- D. M. Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed
- M. Poliakoff, W. Leitner and E. S. Streng, Faraday Discuss., 2015, 183, 9 RSC
-
(a) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC
-
(a) S. Yang, G. S. B. Martin, G. J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374 CrossRef CAS PubMed
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834 CrossRef CAS PubMed
- J. Cavinet, A. Feteeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594 RSC
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724 CrossRef PubMed
- H. A. Lara-García, M. R. Gonzalez, J. H. González-Estefan, P. Sánchez-Camacho, E. Lima and I. A. Ibarra, Inorg. Chem. Front., 2015, 2, 442 RSC
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26, 17198 CrossRef CAS PubMed
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696 RSC
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS
- J. Liu, F. Zhang, X. Zou, G. Yu, N. Zhao, S. Fan and G. Zhu, Chem. Commun., 2013, 49, 7430 RSC
- W. M. Bloch, R. Babaro, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441–10448 CrossRef CAS PubMed
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W.
L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369 CrossRef CAS PubMed
- M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 5472 CrossRef CAS PubMed
-
(a) F. Meunier, Appl. Therm. Eng., 2013, 61, 830 CrossRef
-
(a) S.-i. Noro, R. Matsuda, Y. Hijikata, Y. Inubushi, S. Takeda, S. Kitagawa, Y. Takahashi, M. Yoshitake, K. Kubo and T. Nakamura, ChemPlusChem, 2015, 80, 1517 CrossRef CAS
-
(a) H. Jasuja, Y.-G. Huang and K. S. Walton, Langmuir, 2012, 28, 16874 CrossRef CAS PubMed
-
(a) J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451 CrossRef CAS PubMed
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368 CrossRef CAS PubMed
- E. Soubeyrand-Lenoir, C. Vagner, J. W. Yoon, P. Bazin, F. Ragon, Y. K. Hwang, C. Serre, J.-S. Chang and P. L. Llewellyn, J. Am. Chem. Soc., 2012, 134, 10174 CrossRef CAS PubMed
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606 CrossRef CAS PubMed
-
(a) G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 2976 RSC
- M. Sánchez-Serratos, P. A. Bayliss, R. A. Peralta, E. González-Zamora, E. Lima and I. A. Ibarra, New J. Chem., 2016, 40, 68 RSC
- B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755 CrossRef CAS
-
(a) S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémi, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS PubMed
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75 CrossRef CAS
- P. Serra-Crespo, M. A. van der Veen, E. Gobechiya, K. Houthoofd, Y. Filinchuk, C. E. A. Kirschhock, J. A. Martens, B. F. Sels, D. E. De Vos, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2012, 134, 8314 CrossRef CAS PubMed
- P. Serra-Crespo, A. Dikhtiarenko, E. Stavitski, J. Juan-Alcañiz, F. Kapteijn, F.-X. Coudert and J. Gascon, CrystEngComm, 2015, 17, 276 RSC
- P. A. Bayliss, I. A. Ibarra, E. Pérez, S. Yang, C. C. Tang, M. Poliakoff and M. Schröder, Green Chem., 2014, 16, 3796 RSC
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970 CrossRef CAS PubMed
-
(a) G. R. Medders and F. Paesani, J. Phys. Chem. Lett., 2014, 5, 2897 CrossRef CAS PubMed
- F. Salles, S. Bourrelly, H. Jobic, T. Devic, V. Guillerm, P. Llewellyn, C. Serre, G. Férey and G. Maurin, J. Phys. Chem. C, 2011, 115, 10764 CAS
- I. P. O'koye, M. Benham and K. M. Thomas, Langmuir, 1997, 13, 4054 CrossRef
- J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102 RSC
-
(a) M. R. Gonzalez, J. H. González-Estefan, H. A. Lara-García, P. Sánchez-Camacho, E. I. Basaldella, H. Pfeiffer and I. A. Ibarra, New J. Chem., 2015, 39, 2400 RSC
-
(a) N. L. Ho, F. Porcheron and R. J.-M. Pellenq, Langmuir, 2010, 26, 13287 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: TGA data, PXRD data and kinetic CO2 experiments. See DOI: 10.1039/c5ra26517g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |