DOI:
10.1039/C5RA26493F
(Paper)
RSC Adv., 2016,
6, 11840-11847
Synthesis, coordination behavior and structural features of chiral iron(II) PNP diferrocene complexes†
Received
11th December 2015
, Accepted 16th January 2016
First published on 20th January 2016
Abstract
Five new chiral PNP ferrocene ligands with either an imine or amine nitrogen coordination site were synthesized. Only the imine type ligands formed Fe(II) complexes with the general formula [Fe(PNP)X2] (X = Cl, Br). In the solid state these complexes adopt a tetrahedral geometry with the PNP ligand coordinated in a κ2P,N-fashion with the one pendant-arm and the other not coordinated, as determined by X-ray crystallography and Mössbauer spectroscopy. The complexes are paramagnetic with a quintet ground state. In solution there is an equilibrium between [Fe(κ3P,N,P-PNP)X2] and [Fe(κ2P,N-PNP)X2] complexes. Boronation of the non-coordinated arm shifts the equilibrium towards the four-coordinate complex [Fe(κ2P,N-PNPBH3)Br2]. DFT calculations are consistent with the experimental results and indicate that the experimentally observed κ2 isomer is thermodynamically the most stable. In a CO atmosphere, [Fe(PNP)(CO)2Br]Br was formed rather than [Fe(PNP)(CO)Br2].
Introduction
At present, platinum metal-based asymmetric catalysis is considered to be a standard methodology in organic synthesis and it is frequently used in both academia and industry. In particular, asymmetric hydrogenations on various types of substrates can nowadays be carried out on a very large scale.1–3 However, these processes require rather expensive ruthenium-, rhodium- or iridium-based catalysts.4,5 Not only for the obvious economic reasons, but also driven by the ideas of ‘Green and environmentally friendly chemistry’, it became clear that iron-based catalysts could provide a number of benefits over platinum metal catalysts if they could be made stable, active, selective and easy to handle. In this respect, besides the lower cost and greater abundance of iron, its very low toxicity makes it an attractive candidate for the preparation of well-defined iron-based catalysts.6–14 To date, only one class of efficient chiral PNP iron catalysts for the preparation of enantioenriched alcohols and imines has been reported and this was developed by the Morris group.15 Since, a large number of chiral non-racemic substituted ferrocene derivatives have been synthesized and such planar chiral derivatives have been applied in a variety of different fields, including homogeneous enantioselective catalysis.16 On the basis of the information outlined above, we embarked on a program to synthesize chiral iron complexes based on ferrocene backbones.
The study reported here concerns the synthesis of chiral PNP pincer ligands with a ferrocene scaffold (Chart 1) and the structural behavior and reactivity of their corresponding Fe(II) complexes.
 |
| | Chart 1 | |
Results and discussion
Synthesis of ligand (SFc,SFc)-1 and (SFc,SFc)-2
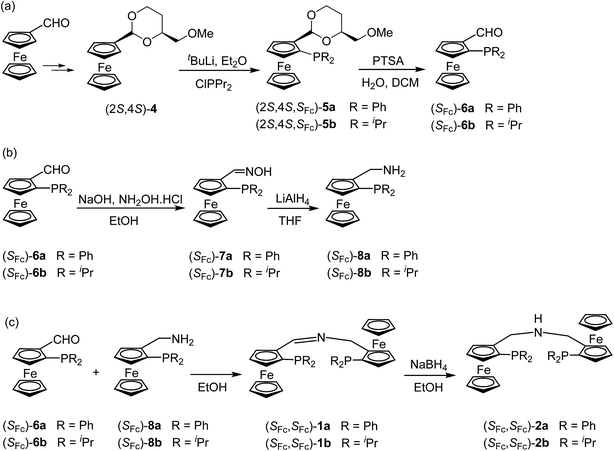
As outlined in Scheme 1, enantiopure aldehyde (SFc)-6, which was prepared by applying Kagan's methodology,17 was reacted with sodium hydroxide and hydroxylamine hydrochloride to give (SFc)-7. Subsequent treatment with LiAlH4 in THF gave (SFc)-8.18 In the final step, condensation of (SFc)-6 and (SFc)-8 in ethanol yielded the desired ligands (SFc,SFc)-1. The molecular structure of (SFc,SFc)-1a was determined by X-ray diffraction (Fig. 1). Treatment of (SFc,SFc)-1 with NaBH4 in ethanol gave (SFc,SFc)-2 in quantitative yield (Scheme 1).
 |
| | Scheme 1 | |
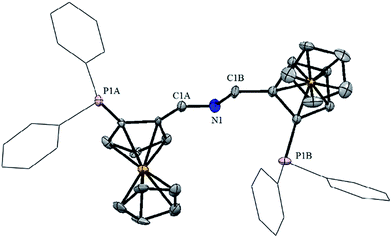 |
| | Fig. 1 Structural view of (SFc,SFc)-1a showing 50% thermal ellipsoids (H atoms and solvent molecules are omitted for clarity). Selected bond lengths (Å) and bond angles (°): C1A–N1 1.29(1), C1B–N1 1.40(1), C1A–N1–C1B 117.5(8). | |
Synthesis of ligand (R,R,SFc,SFc)-3
(R,SFc)-10, which is easily accessible from (R)-Ugi's amine ((SFc)-9), was reacted with Ac2O at 100 °C for 2 hours to afford (R,SFc)-11 followed by treatment with a large excess of ammonia in ethanol in an autoclave at 80 °C for 6 hours to yield the primary amine (R,SFc)-12.19 A mixture of (R,SFc)-9 and (R,SFc)-12 in acetonitrile and water was heated under reflux for 30 hours to give (R,R,SFc,SFc)-3 in 61% isolated yield (Scheme 2).
 |
| | Scheme 2 | |
Synthesis of iron complexes
Treatment of (SFc,SFc)-1 with 0.97 equivalents of FeBr2(THF)2 (ref. 20) or anhydrous FeCl2 in THF gave the coordinatively unsaturated complexes (SFc,SFc)-13a, (SFc,SFc)-13b, and (SFc,SFc)-13c in 93%, 80% and 91% yield, respectively (Scheme 3). The molecular structures of (SFc,SFc)-13a and (SFc,SFc)-13c were determined by X-ray crystallography and selected bond distances and angles are given in the captions (Fig. 2 and 3). Both structures have a distorted tetrahedral geometry around the Fe(II) center, with the PNP ligand coordinated in a κ2P,N-fashion rather than in the typical κ3P,N,P-coordination mode. This bonding mode is relatively rare for pincer type ligands. We recently described a taddol based Fe(II) PNP pincer complex with a 2,6-diaminopyridine backbone where the ligand was also coordinated in a κ2P,N-fashion.21 The Fe–P bond length of about 2.4 Å is comparable with those in other high-spin tetrahedral iron(II) complexes.22
 |
| | Scheme 3 | |
 |
| | Fig. 2 Structural view of (SFc,SFc)-13a showing 50% thermal ellipsoids (H atoms and solvent molecules are omitted for clarity). Selected bond lengths (Å) and bond angles (°): Fe1–Br1 2.3818(5), Fe1–Br2 2.3916(5), Fe1–P1 2.3999(9), Fe1–N1 2.090(3), P1–Fe1–Br1 110.42(3), P1–Fe1–N1 95.16(8), Br1–Fe1–N1 115.47(8), Br2–Fe1–P1 112.84(3), Br2–Fe1–N1 102.08(8). | |
 |
| | Fig. 3 Structural view of (SFc,SFc)-13c showing 50% thermal ellipsoids (H atoms and solvent molecules are omitted for clarity). Selected bond lengths (Å) and bond angles (°): Fe1–Cl1 2.2438(8), Fe1–Cl2 2.2472(8), Fe1–P1 2.4031(8), Fe1–N1 2.095(2), Cl1–Fe1–Cl2 119.83(3), P1–Fe1–Cl1 110.96(3), P1–Fe1–Cl2 112.63(3), Cl1–Fe1–N1 113.32(7), Cl2–Fe1–N1 106.64(7). | |
Although better coordination was expected for the ligand (SFc,SFc)-1b which contains more strongly electron donating PiPr2 moieties, similar results to (SFc,SFc)-1a were obtained and also this ligand is coordinated in κ2P,N-fashion. On the other hand, the ligands (SFc,SFc)-2 and (R,R,SFc,SFc)-3, under analogous reaction conditions, did not react with the iron salts and coordination did not take place. The reason for this behavior is not clear but it could be due to the larger bite angles in these ligands. As a consequence, the following discussion is focused on complexes obtained from (SFc,SFc)-1a.
Mössbauer spectroscopy
The magnetic properties of (SFc,SFc)-13a were investigated by means of Mössbauer spectroscopy. The Mössbauer spectrum of complex (SFc,SFc)-13a at 78 K is well defined and was fitted with three different iron sites (Fig. 4). The blue and orange sites were present in equal proportions (32.9%) and their quadrupole splitting (QS) values obtained from the fitting are very similar. The isomer shift (IS) values are slightly different, i.e., 0.47(1) and 0.55(1) mm s−1, and the Fe site with the lowest IS corresponds to the one with the highest s-electron density at the nucleus. Globally, the hyperfine parameters obtained for these two sites are reminiscent of the ones for Fe(II) in ferrocene (Table 1),23 a finding that is consistent with the structure determined by X-ray crystallography. For the third site (green) the expected proportion (34.2%) obtained from the fit shows that it corresponds to the central iron in a distorted tetrahedral coordination environment. The IS value is consistent with HS Fe(II) (Table 1).
 |
| | Fig. 4
57Fe Mössbauer spectrum of [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a collected at 78 K. | |
Table 1 Estimated hyperfine parameters from the 57Fe Mössbauer spectra of (SFc,SFc)-13a collected at 78 K and related tetrahedral Fe(II) complexes. IS: isomer shift; QS: quadrupole splitting
| Complex |
IS (mm s−1) |
QS (mm s−1) |
Fe(II) |
Ref. |
| |
| [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a |
0.782(1) |
3.208(2) |
HS |
This work |
| 0.467(3) |
2.203(3) |
LS (FeCp2 arm) |
| 0.552(3) |
2.190(3) |
LS (FeCp2 arm) |
| Ferrocene |
0.531(1) |
2.491(1) |
LS |
23
|
| [Fe(κ2P,N-PNP-iPr,TAD)Cl2] |
0.78(1) |
2.93(1) |
HS |
21
|
| [Fe(κ2P,N-PNP-iPr,TAD)Br2] |
0.76(1) |
3.14(1) |
HS |
21
|
| [Fe(κ2P,N-PNP-tBu,TAD)Cl2] |
0.79(1) |
3.06(1) |
HS |
21
|
| [Fe(κ2P,N-PNP-tBu,TAD)Br2] |
0.77(1) |
3.22(1) |
HS |
21
|
| [Fe(SNH-Ph)Br2] |
0.855(1) |
3.282(2) |
HS |
32
|
| [Fe(SNEt-Ph)Cl2] |
0.899(1) |
3.008(1) |
HS |
32
|
| [Fe(SNEt-Ph)Br2] |
0.877(1) |
3.259(2) |
HS |
32
|
| [Fe(PNH-iPr)Cl2] |
0.680(2) |
2.871(4) |
HS |
33
|
| [Fe(SNH-iPr)Cl2] |
0.796(2) |
2.998(4) |
HS |
33
|
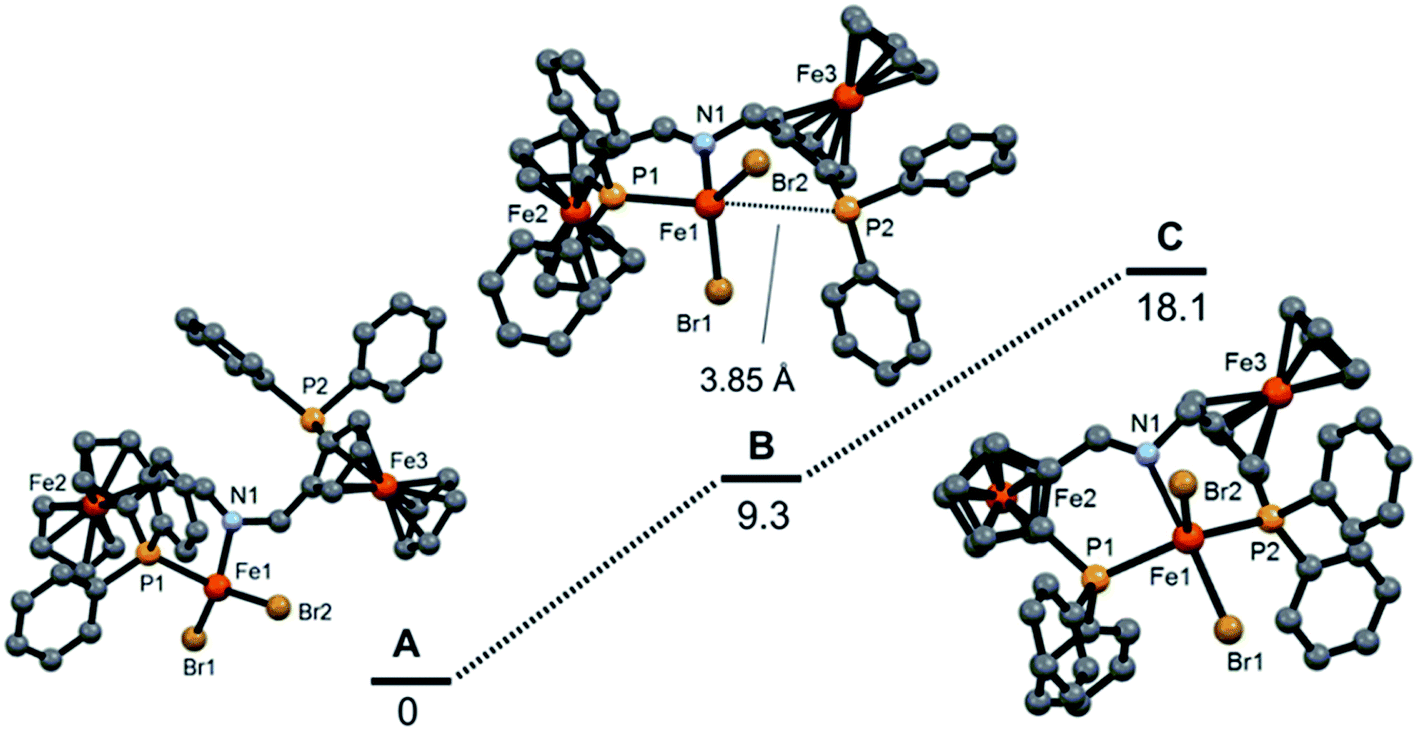
DFT calculations for complex [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a are in agreement with the experimental data and this indicates that the experimentally observed isomer (denoted as A) is thermodynamically more stable by 9.3 and 18.1 kcal mol−1, respectively, than the isomers B and C (Fig. 5), which were not observed. In A and B one phosphine moiety is not coordinated and B is a rotamer of A with a long Fe⋯P distance of 3.85 Å. Complex C is a five-coordinate species with a distorted square pyramidal geometry.
 |
| | Fig. 5 Free energies (B3LYP) of optimized iron PNP complexes featuring κ2P,N and κ3P,N,P-bound PNP ligands. The free energy values (kcal mol−1) are referred to the experimentally obtained tetrahedral [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a (A). | |
Solution studies
The structures of complexes (SFc,SFc)-13a and (SFc,SFc)-13c in the solid state are were unequivocally confirmed by X-ray crystallography, but in solution all of the complexes gave 1H and 13C{1H} NMR spectra with large paramagnetic shifts. The signals were broad and featureless and, due to the complexity of the PNP ligands it was not possible to assign signals, thus making the spectra very uninformative. The solution magnetic moment of the dibromide (SFc,SFc)-13a was measured to be 4.98μB (Evans' method). This value is in good agreement with the effective magnetic moment of HS Fe(II) in the spin only approximation (4.9μB). Accordingly, it is not clear whether these complexes are four- or five-coordinate and contain κ2P,N- or κ3P,N,P-bound PNP ligands, respectively, or whether both species are present and, if this is the case, whether these are in equilibrium with one another. As a consequence, further experiments were carried out in an effort to elucidate the structure of (SFc,SFc)-13a in solution. At room temperature 31P{1H} NMR signals could not be detected at all, but at −80 °C a comparatively sharp signal was observed at around 118 ppm and, this was assigned to the non-coordinated arm of the PNP ligand. This observation allows the existence of equilibria between κ2P,N- and κ3P,N,P-bound species to be postulated. Moreover, the addition of BH3·THF to a solution of (SFc,SFc)-13a in CD2Cl2 led to boronation of the non-coordinated arm and a shift in the equilibrium towards the four-coordinate complex [Fe(κ2P,N-PNPBH3)Br2] (SFc,SFc)-14 (Scheme 4). The 31P{1H} NMR spectra obtained in these studies are shown in Fig. 6. The molecular structure of (SFc,SFc)-14 was determined by X-ray crystallography (Fig. 7) and this confirmed the pendant phosphorus moiety had been boronated.
 |
| | Scheme 4 | |
 |
| | Fig. 6
31P{1H} NMR spectra of (a) (SFc,SFc)-1a, (b) [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a at −80 °C, and (c) [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-14 in CD2Cl2 (spectra of (SFc,SFc)-13a and (SFc,SFc)-14 are referenced internally to PPh3 set to −5.6 ppm). | |
 |
| | Fig. 7 Structural view of (SFc,SFc)-14 showing 50% thermal ellipsoids (H atoms and solvent molecules are omitted for clarity). Selected bond lengths (Å) and bond angles (°): Fe1–Br1 2.3911(7), Fe1–Br2 2.3887(6), Fe1–P1 2.400(1), Fe1–N1 2.088(3), P2–B1 1.927(5), Br1–Fe1–Br2 117.74(3), P1–Fe1–Br1 110.70(3), P1–Fe1–Br2 112.80(3), Br1–Fe1–N1 115.53(9), Br2–Fe1–N1 102.68(9). | |
Reactivity towards CO
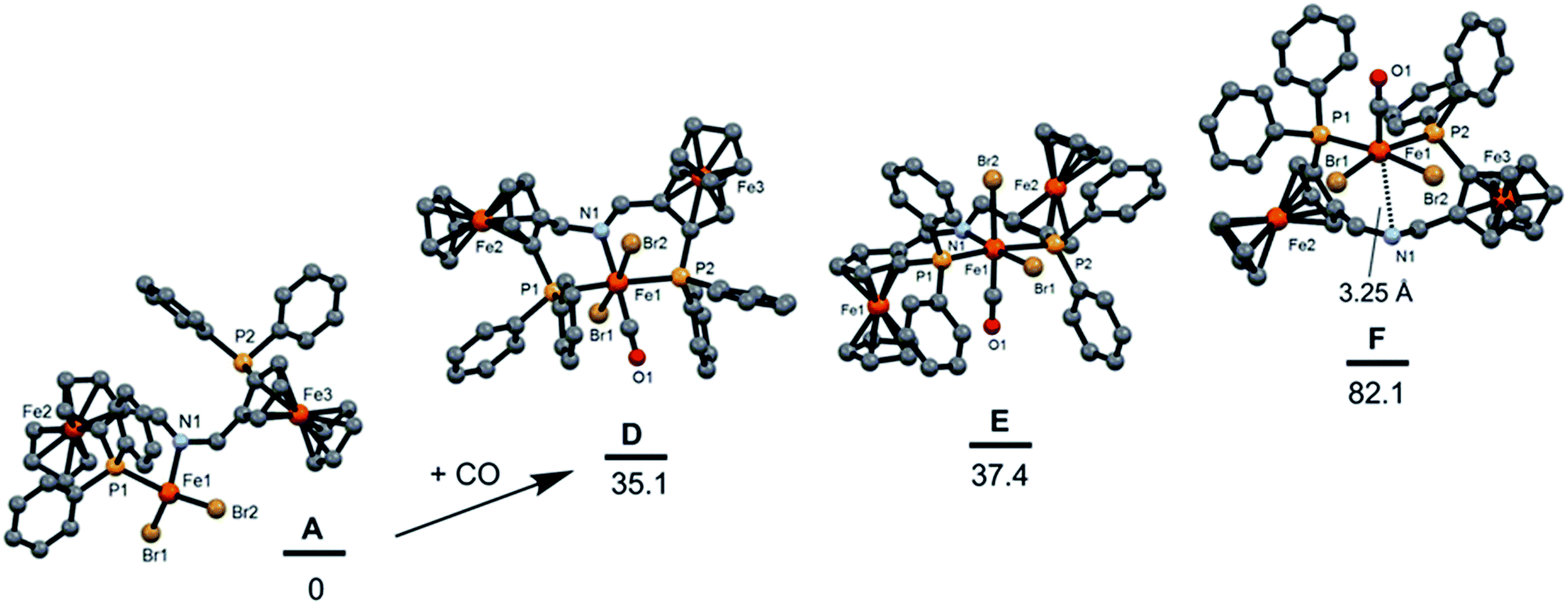
In order to obtain iron-based active hydrogenation catalysts it appears to be important to have diamagnetic complexes. Accordingly, virtually all iron complexes that are active catalysts feature the strong field ligand CO, which seems to maintain a low spin configuration throughout the catalytic cycle.24–31 Therefore, (SFc,SFc)-13a was stirred overnight in CH2Cl2 under a CO atmosphere (2 bar) in an autoclave to obtain mono-CO or bis-CO complexes of the types (SFc,SFc)-15a and (SFc,SFc)-16a (Scheme 5). Under these conditions mono-CO complexes could not be isolated and only (SFc,SFc)-16a was obtained. Due to the fact that CO compounds are not stable and readily release CO, NMR measurements on (SFc,SFc)-16a were carried out under a CO headspace. The 31P{1H} NMR spectrum contained two major and three minor pairs of doublets with 2Jpp values in the range 165–178 Hz. These signals are indicative of a trans-phosphorus arrangement. The 1H NMR spectrum was not very informative due to its complexity and the presence of isomers. The HRMS revealed a fragment of [Fe(PNP)(CO)2Br]+ ([M]+) at m/z 969.9684. The IR spectra displayed two strong νCO absorptions at 1990 and 2038 cm−1, which implies a cis-CO geometry around the iron center. However, attempts to grow crystals under a CO atmosphere were unsuccessful. The experimental results are in agreement with DFT calculations, where bis carbonyl complexes (SFc,SFc)-16a, in which the phosphorus moieties are in a mutual trans position, are more stable by 50 kcal mol−1 than the respective complexes (SFc,SFc)-17a, which have a cis phosphorus arrangement (Scheme 5). In accordance with these results, DFT calculations revealed that addition of CO to (SFc,SFc)-13a is a thermodynamically highly unfavorable process (Fig. 8). The putative CO complexes trans-P, trans-Br-[Fe(PNP)(CO)Br2] (D), cis-Br, trans-P-[Fe(PNP)(CO)Br2] (E) and cis-Br, cis-P-[Fe(PNP)(CO)Br2] (F) are 35.1, 37.4, and 88.9 kcal mol−1, respectively, are less stable than the initial reagent A (SFc,SFc)-13a.
 |
| | Scheme 5 | |
 |
| | Fig. 8 Free energy profile (B3LYP) for the addition of CO to form the illusive complexes trans-P, trans-Br-[Fe(PNP)(CO)Br2] (SFc,SFc)-15a (D), cis-Br, trans-P-[Fe(PNP)(CO)Br2] (SFc,SFc)-15a (E) and cis-Br, cis-P-[Fe(PNP)(CO)Br2] (SFc,SFc)-15a (F). The energy values (kcal mol−1) are referred to [Fe(κ2P,N-PNP)Br2] (SFc,SFc)-13a (A). | |
In addition, (SFc,SFc)-16a was also prepared from Fe(CO)4(Br)2, as a different iron source, and the ligand (SFc,SFc)-1a and the structure was established by NMR, HRMS and IR. In a similar way (SFc,SFc)-16b was prepared from (SFc,SFc)-13a and this compound had comparable spectroscopic data to (SFc,SFc)-16a.
In the next step, in an attempt to remove a bromide ligand, (SFc,SFc)-16a and (SFc,SFc)-16b were reacted with 1.07 equivalents of a halide abstractor (AgBF4 and AgPF6) in THF under a CO atmosphere (2 bar). The inclusion of a halide abstractor led to an immediate color change from red to red-purple. The 31P{1H} NMR spectra of isolated compounds showed the formation of a new compound with an ABX pattern and 2JPP couplings in the range 44–47 Hz. However, the mixtures were intractable and all attempts to characterize these compounds failed.
Synthesis of hydride complexes
Iron hydride complexes as active catalysts for the asymmetric hydrogenation of ketones and imines were synthesized by treating iron dicarbonyl complexes (SFc,SFc)-16a and (SFc,SFc)-16b with various hydride reagents such as Na[HBEt3], NaBH4 and LiAlH4. However, in all cases decomposition was observed and the main product was the reduced free ligand. As mentioned above, the cases of (SFc,SFc)-2 and (R,R,SFc,SFc)-3, neither of which contain an imine bond, coordination with iron was not observed. This finding could explain the failure of all attempts to form iron hydride complexes and the fact that the free ligand was obtained.
Catalysis
(SFc,SFc)-16a and (SFc,SFc)-16b were used as catalyst precursors in the asymmetric hydrogenation of acetophenone (reaction conditions: 25 °C, 5 bar H2, 0.1 mol% of precatalyst and 4% potassium tert-butoxide). However, it was found that these compounds are not active in catalysis.
Conclusions
New chiral imine and amine type PNP ligands, all based on diferrocene scaffold, were developed. While the amine type ligands did not coordinate to iron with use of the imine ligands Fe(II) complexes of general formula [Fe(PNP)X2] (X = Cl, Br) were obtained. In the solid state, as established by X-ray crystallography and supported by Mössbauer result, these complexes adopt a tetrahedral geometry with the PNP ligand coordinated in a κ2P,N-fashion with one pendant-arm but the other arm is not coordinated. All complexes are paramagnetic with a quintet ground state. Equilibria exist in solution between [Fe(κ3P,N,P-PNP)X2] and [Fe(κ2P,N-PNP)X2] complexes. Boronation of the non-coordinated arm shifts the equilibrium towards the four-coordinate complex [Fe(κ2P,N-PNPBH3)Br2]. The results of DFT calculations are in agreement with the experimental results and indicates that the experimentally observed κ2P,N isomer is thermodynamically the most stable. Under a CO atmosphere [Fe(PNP)(CO)2Br]Br was formed rather than [Fe(PNP)(CO)Br2]. However, this complex decomposed upon removing the CO atmosphere and [Fe(PNP)Br2] was formed. All attempts to transform [Fe(PNP)(CO)2Br]Br complexes into hydride derivatives resulted in decomposition of complexes and gave the reduced ligand as a main product. The reluctance of the Fe(PNP) complexes to form low-spin mono CO complexes, which seems to be a prerequisite for the subsequent formation of hydride complexes, may explain, why no hydride complexes are formed and thus why these complexes are inactive in hydrogenation catalysis. All complexes remain in the high spin state and renders the PNP ligands too labile, which upon treatment with hydride sources liberate the PNP ligands. This again shows that small electronic changes in the ligand environment of first row transition metal complexes has a drastic impact on reactivity.
Computational details
Calculations were performed using the GAUSSIAN 09 software package, and the B3LYP functional without symmetry constraints. This functional was shown to perform well in mechanistic studies of spin forbidden reactions in closely related Fe systems. The optimized geometries were obtained with the Stuttgart/Dresden ECP (SDD) basis set to describe the electrons of the iron atom. For all other atoms a standard 6-31G** basis set was employed. Frequency calculations were performed to confirm the nature of the stationary points the yielded no imaginary frequency for the minima.
Acknowledgements
Financial support from the Austrian Science Fund (FWF) is gratefully acknowledged (Projects No. T 631-N28). The authors wish to express sincere appreciation and gratitude to Professor Walter Weissensteiner for his kind support and helpful discussions and comments during the course of this research. The NMR Center the University of Vienna – in particular Susanne Felsinger – is acknowledged. The X-ray center of the University of Vienna and Vienna University of Technology is also acknowledged for financial support and for providing access to the single-crystal diffractometer. MDC and LPF acknowledge Fundação para a Ciência e Tecnologia (Projects UID/MULTI/00612/2013 and UID/MULTI/04046/2013).
References
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- F. Naud, F. Spindler, C. J. Rueggeberg, A. T. Schmidt and H.-U. Blaser, Org. Process Res. Dev., 2007, 11, 519–523 CrossRef CAS.
-
Handbook of Homogeneous Hydrogenation, ed. J. G. De Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, vol. 40, pp. 1–3 Search PubMed.
-
I. Ojima, Catalytic Asymmetric Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
-
H. Brunner and W. Zettlmeier, Handbook of Enantionselective Catalysis, VCH, Weinheim, 1993 Search PubMed.
- P. Bhattacharya and H. Guan, Comments Inorg. Chem., 2011, 32, 88–112 CrossRef CAS.
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341–1369 CrossRef CAS.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- L.-X. Liu, Curr. Org. Chem., 2010, 14, 1099–1126 CrossRef CAS.
- M. Zhang and A. Zhang, Appl. Organomet. Chem., 2010, 24, 751–757 CrossRef CAS.
- K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- H. Nakazawa and M. Itazaki, Organomet. Chem., 2011, 33, 27–81 CAS.
- P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367–1380 CrossRef CAS PubMed.
-
P. Štěpnička, Ferrocenes: Ligands, Material and Biomolecules, Wiley, Chichester, 2008 Search PubMed.
- O. Riant, O. Samuel, T. Flessner, S. Taudien and H. B. Kagan, J. Org. Chem., 1997, 62, 6733–6745 CrossRef CAS.
- X. Hu, H. Dai, X. Hu, H. Chen, J. Wang, C. Bai and Z. Zheng, Tetrahedron: Asymmetry, 2002, 13, 1687–1693 CrossRef CAS.
- W. Li, T. Wei, Y. Gao, K. Xi and X. Jia, Polymer, 2012, 53, 1236–1244 CrossRef CAS.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H. J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- C. Holzhacker, B. Stöger, D. M. Carvalho, L. P. Ferreira, E. Pittenauer, G. Allmaier, L. F. Veiros, S. Realista, A. Gil and M. J. Calhorda, Dalton Trans., 2015, 44(29), 13071–13086 RSC.
- E. J. Hawrelak, H. Wesley, E. Lobkovsky, G. T. Yee, E. Bill and P. Chirik, Inorg. Chem., 2005, 44(9), 3103–3111 CrossRef CAS PubMed.
- H. Schottenberger, M. R. Buchmeiser and R. H. Herber, J. Organomet. Chem., 2000, 612, 1–8 CrossRef CAS.
- P. O. Lagaditis, P. E. Sues, A. J. Lough and R. H. Morris, Dalton Trans., 2015, 44, 2119–12127 RSC.
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 4111 CAS.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed.
- R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2012, 18, 7196–7209 CrossRef CAS PubMed.
- C. Holzhacker, M. J. Calhorda, A. Gil, M. D. Carvalho, L. P. Ferreira, K. Mereiter, B. Stöger, E. Pittenauer, G. Allmaier and K. Kirchner, Polyhedron, 2014, 81, 45–55 CrossRef CAS.
- C. Holzhacker, C. M. Standfest-Hauser, M. Puchberger, K. Mereiter, L. F. Veiros, M. J. Calhorda, M. D. Carvalho, L. P. Ferreira, M. Godinho, F. Hartl and K. A. Kirchner, Organometallics, 2011, 30, 6587–6601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 31P{1H} NMR spectra of all ligands and complexes, (SFc,SFc)-1a, (SFc,SFc)-13a, (SFc,SFc)-13c, and (SFc,SFc)-14. CCDC 1435577, 1435448, 1433174 and 1433175. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra26493f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a