
Palladium-catalyzed direct alkenylation of 4-hydroxy-2-pyridones†
Abstract
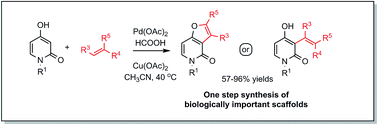
The first direct C-3 alkenylation of N-substituted-4-hydroxy-2-pyridones with unactivated alkenes has been achieved under conventional palladium acetate catalysis. The presented protocol enables the efficient production of functionalized furo[3,2-c]-pyridones-2 when terminal alkenes are utilized and 3-alkenyl-4-hydroxy-2-pyridones when more substituted reaction partners are involved. The mild reaction conditions allow easy, scalable access to pyridone derivatives that resemble core structures isolated from natural alkaloids.
 Please wait while we load your content...
Please wait while we load your content...

