
PEG/heparin-decorated lipid–polymer hybrid nanoparticles for long-circulating drug delivery
Yan Sheng*a,
Lingqian Chang*b,
Tairong Kuangb and
Jiaming Hub
aCollege of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, People's Republic of China. E-mail: sh-crystal@163.com
bNanoscale Science and Engineering Center, The Ohio State University, Columbus, OH 43210, USA. E-mail: chang.982@osu.edu
First published on 25th February 2016
Abstract
The clinical success of lipid–polymer hybrid nanoparticles (LPHNPs) for effective targeted drug delivery is still hindered by their rapid clearance from the bloodstream. In this work, a novel strategy for surface modification of LPHNPs with combined polyethylene glycol (PEG) and heparin (HEP) was developed to achieve a significant prolongation in blood circulation. All the LPHNPs formulated with a diameter of 100–200 nm were prepared by a modified w/o/w solvent diffusion/evaporation method and physicochemically characterized. The synergistic action of PEG and HEP was observed, as combinatorial modification significantly improved the surface hydrophilicity as well as the suspension stability of nanoparticles and tailored the surface charge close to neutrality, in comparison to LPHNPs surface-treated with PEG or HEP alone. In vitro and in vivo studies showed that the PEG/HEP coating significantly prohibited the macrophage uptake and extended the blood circulation of LPHNPs with concomitant reduced liver sequestration. The in vitro phagocytosis results using murine peritoneal macrophages showed 8.2-fold reduction compared to the control LPHNP group. The in vivo study in ICR mice showed PEG/HEP coating increased the blood circulation half-life of LPHNPs from 0.3 h to 72.6 h. Moreover, PEG/HEP LPHNPs exhibited dramatically reduced liver accumulation when compared to LPHNPs. These results demonstrated that PEG/HEP LPHNPs with optimized particle size, surface hydrophilicity and surface charge, have a promising potential as long-circulating drug delivery systems.
Introduction
Lipid–polymer hybrid nanoparticles (LPHNPs) have emerged as a promising drug delivery system for cancer therapy recently.1–3 With complementary characteristics of both liposomes and polymeric nanoparticles, LPHNPs exhibit biocompatibility, biodegradability, high structural integrity, excellent encapsulation efficiency and controlled drug release. However, a major barrier for effective delivery of various chemotherapeutic drugs to the target site is the sequestration of intravenously administrated LPHNPs by cells of the mononuclear phagocytic system (MPS), and then quick elimination from the systemic circulation.4 It is well-accepted that adsorption of plasma proteins (opsonins) onto the particle surface, also known as opsonization, is critical to trigger the phagocytic uptake.5,6 Therefore, an ‘invisible’ surface that evades opsonin adsorption and subsequent clearance from the blood by phagocytic cells is the key factor to achieve long-circulating LPHNPs.Physicochemical properties of nanoparticles, including particle size, surface charge and surface chemistry, are key parameters determining their biological fate after intravenous (i.v.) administration.7 The desired diameter of intravascular long-circulating nanoparticles has been reported within the size range of 70–200 nm.8,9 A successful strategy for camouflaging nanoparticles is the use of surface adsorbed or grafted shielding groups. Representatively, such surface groups include polyethylene glycol (PEG), polysaccharides and poloxamers, etc. Among them, PEG coating has been extensively studied on a variety of nanoparticle systems to prolong circulation time by decreasing interactions with blood proteins and MPS cells.10–12 Polysaccharides have been recognized as a promising material for developing long-circulating systems. Hydrophilic polysaccharide coatings, such as heparin (HEP), dextran and water-soluble chitosan, have been reported to decrease the phagocytic uptake of particulate carriers.13–16 The in vivo circulation half-life (t1/2) of nanoparticles can be significantly prolonged from several minutes to hours by surface modification with the single-component coating.14,17,18 However, considering the solid-tumor treatment, the blood survival time of these nanoparticles in the body could not achieve effective drug targeting and therapy.
To address this, our idea in here was to develop a new class of combinatorial coating of PEG and HEP for the sake of effectively improving the prolongevity of LPHNPs in blood. As schematized in Scheme 1, polylactic acid (PLA) was chosen as a model polymer serving as the shell for drug core, while, PEG and HEP cooperatively formed the corona of the nanoparticles, and 1,2-di-O-octadecenyl-3-trimethylammonium propane (chloride salt) (DOTMA), as a model cationic lipid, formed the lipid monolayer at the interface of the PLA shell and PEG/HEP corona. Bare LPHNPs and LPHNPs modified with PEG and HEP alone were also included for comparison. The influences of different surface coatings on the surface hydrophilicity and surface charge of nanoparticles were compared. The biological behaviors of these LPHNPs labeled with coumarin-6 were investigated in terms of blood clearance and biodistribution following i.v. administration in ICR mice. To have a better understanding of their in vivo elimination by MPS system, the in vitro phagocytic uptake assay was also studied. In this work, DOX hydrochloride salt (DOX) was selected as a model cancer drug.
 | ||
| Scheme 1 Schematic illustration of PEG/HEP decorated LPHNPs containing DOX. The DOX-loaded PEG/HEP LPHNPs display a core–shell–corona structure. The hydrophobic PLA shell serves as the reservoir for DOX core, while the hydrophilic PEG and heparin as the corona. A cationic lipid DOTMA monolayer was formed at the interface of the shell and the corona. | ||
Experimental
Materials
DOTMA was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). HEP sodium salt, derived from porcine mucosa (Mw 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), was obtained from Wako Pure Chemical Industries (Osaka, Japan). PLA (Mw 40k) and PLA–PEG diblock copolymer (Mw 110k, MwPLA:MwPEG = 80:20) were supplied by DaiGang Biotechnology Co. Ltd., Jinan, China. DOX was obtained from Beijing Huafeng United Technology Co. (China). ICR mice were provided by Experimental Animal Center of Shandong Luye Pharmaceutical Co. Ltd., Yantai, China. Ultrapure water (Millipore, Bedford, MA, USA) was used throughout the experiment. All other reagents and solvents were of analytic grade.
000), was obtained from Wako Pure Chemical Industries (Osaka, Japan). PLA (Mw 40k) and PLA–PEG diblock copolymer (Mw 110k, MwPLA:MwPEG = 80:20) were supplied by DaiGang Biotechnology Co. Ltd., Jinan, China. DOX was obtained from Beijing Huafeng United Technology Co. (China). ICR mice were provided by Experimental Animal Center of Shandong Luye Pharmaceutical Co. Ltd., Yantai, China. Ultrapure water (Millipore, Bedford, MA, USA) was used throughout the experiment. All other reagents and solvents were of analytic grade.
Preparation of DOX-loaded LPHNPs
DOX-loaded LPHNPs were prepared by a w1/o/w2 solvent diffusion/evaporation method19 with some modifications. 0.5 mL DOX solution (2 mg mL−1) (w1 phase) was emulsified in 5 mL of organic solvent phase (o phase) containing PLA-based polymer (7 mg), DOTMA (3 mg) and 6-coumarin (10 μg) by ultrasonic (JYD-900, Zhixin Instrument Co., Ltd., Shanghai, China) for 15 s. Thereafter, the primary emulsion was poured into 50 mL 0.5% poly(vinyl alcohol) (PVA) aqueous solution (w2 phase) followed by two steps of re-emulsification by a high-pressure homogenizer (AH110D, ATS Engineering Inc., Brampton, Canada) to obtain the double emulsion. The double emulsion was subsequently dispersed into 150 mL 0.5% PVA aqueous solution (w2 phase) and then vacuumed to completely remove the solvents. The LPHNPs were collected by centrifugation (GL-21 M, Shanghai Centrifuge Institute Co., Ltd., Shanghai, China) at 20000g for 60 min, followed by washing three times with Millipore water before lyophilization. The entire process was maintained at 4 °C by thermostatted water bath.
Surface modification of LPHNPs
For the preparation of LPHNPs coated with PEG alone (PEG LPHNPs), PEG was co-incorporated into the nanoparticle matrix as copolymer. In this way, 7 mg PLA–PEG was dissolved in the o phase during the nanoparticle formulation process. For the preparation of LPHNPs coated with HEP alone (HEP LPHNPs), 7 mg PLA was dissolved in the o phase as matrix polymer and 0.1% HEP was added in the w2 phase as modifier. For the preparation of nanoparticles coated with the combined PEG and HEP (PEG/HEP LPHNPs), 7 mg PLA–PEG was dissolved in the o phase as matrix polymer and 0.1% HEP was added in the w2 phase as modifier.Characterization of LPHNPs
The morphology of the freeze-dried nanoparticle powders was observed under scanning electron microscopy (SEM) (S-4800, HITACHI, Japan). Samples were transferred onto a small metal cylinder and coated with gold prior to SEM imaging.Particle size distribution and zeta potential values of the prepared LPHNPs were determined at 25 °C by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instrument Ltd., UK). For measurements, samples were diluted to the appropriate concentration with Millipore water and dispersed using ultrasonic baths.
Hydrophilicity of nanoparticle surface was evaluated by measuring the static contact angle of particle film.20 Briefly, nanoparticle suspension (1 mg mL−1 Millipore water) was spin-coated onto a cleaned glass slide (100 × 100 × 1 mm3) at 1500 rpm for 45 s. Thereafter, the glass slides were allowed to dry at 60 °C for 24 h to remove the dispersion medium for the formation of the particle film. In each experiment, a drop of deionized water (5 μL) was added on the glass slide sample and the contact angle was measured at 25 °C by directly reading from the contact angle goniometer (OCA20, Germany). Each value is the average of five measurements conducted on different sites of the sample surface.
The encapsulation efficiency (EE), indicating the loading capacity of drugs, was determined by measuring the DOX concentration in the supernatant using a UV-vis spectrophotometer (U-2001, HITACHI, Japan) at 485 nm.21 The EE was calculated from the following equation:
| EE (%) = (W0 − Wt)/W0 × 100% |
In vitro drug release profiles
The profiles for the in vitro release of DOX from nanoparticles were obtained by dialysis of the DOX-loaded nanoparticle suspensions in phosphate-buffered saline (PBS) (pH 7.4). Drug release was carried out at 37 °C in a water bath with magnetic stirring (100 rpm). At defined intervals, release medium was collected and the amount of DOX released from the nanoparticles was analyzed by spectrophotometry at a wavelength of 485 nm.In vitro suspension stability of LPHNPs
Since these LPHNPs were developed as carriers for i.v. administration of drugs, it is necessary to evaluate their suspension stability in a physiological medium. The suspension stability assay was performed on the basis of turbidity measurements using an optical analyzer Turbiscan (Formulaction, France) as previously described by our group.22 In brief, 5 mL of nanoparticle suspension was incubated in 5 mL of fetal bovine serum (FBS) (SiJiQing Biomedical Inc., Hangzhou, China) for 120 h at 37 °C. During incubation, a light beam emitted in near infrared (880 nm) wavelength scanned the nanoparticle suspension at each predetermined time point. Two synchronous optical sensors received light transmitted through the sample and backscattered by the sample, respectively. The variation of backscattering data (ΔBS) was directly dependent on the particle size distribution, which was used to assess the stability of nanoparticle suspension.Cytotoxicity studies
The cytotoxicity of LPHNPs with different coatings was assessed by a MTT viability assay against human vascular endothelial cells (EVC-304 cells). Cells were seeded at 30000 cells per well in complete DMEM supplemented with 10% FBS followed by incubation at 37 °C in a 5% CO2 incubator for 24 h. The cytotoxicity was evaluated by determining the viability of the cells after incubation with different concentrations of LPHNPs, PEG LPHNPs, HEP LPHNPs or PEG/HEP LPHNPs (from 0.0625 mg mL−1 to 1 mg mL−1) for 48 h. The number of viable cells was determined by the estimation of their cellular reductive capacity to metabolise the yellow tetrazolium salt (MTT) to a highly colored formazan product. At the end of the incubation period with LPHNPs, cells were incubated with 100 μL of a MTT solution (5 mg mL−1) for 4 h at 37 °C. One hundred microliters of DMSO were then added in order to dissolve the formazan crystals. The UV absorbance of the solubilized formazan crystals was measured spectrophotometrically (MULTISKAN MK3, Thermo Electron Corporation, USA) at 490 nm. Cell viability was expressed as the ratio between the amount of formazan determined for cells treated with the different nanoparticle suspensions and for control non-treated cells. Each point was performed in quadruplicate.
In vitro phagocytosis test
In vitro phagocytic uptake of LPHNPs was carried out using elicited mouse peritoneal macrophages (MPMs) harvested from ICR mice (25 ± 5 g) by intraperitoneal injection of 1 mL 2.9% thioglycolate medium (Sigma Chemical Co.). Cells were isolated by centrifugation at 2000 rpm for 10 min and then re-suspended in complete culture medium. Thereafter, the cells were seeded into 24-well Costar® culture plates (Corning, NY, USA) at a density of 1.3 × 105 cells per cm2, incubating in a humidified atmosphere containing 5% CO2 at 37 °C for 6 h. Thereafter, the purification was performed by washing with PBS (pH 7.4) to remove non-adherent cells and then culture under the same conditions until they formed a confluent monolayer. Thereafter, 100 μL of coumarin-6 labeled nanoparticle suspension (200 μg mL−1 in HBSS) were incubated with the MPM at 37 °C for 1 h. Then, the cells were washed five times with ice PBS to remove the particles adhered to the cell surface. The phagocytized particles were observed using a fluorescence microscope (Nikon, USA). To quantify the phagocytized nanoparticles, 300 μL of 10% Triton X-100 in acetonitrile solution was added in each well to expose the internalized nanoparticles. The fluorescent intensity of cell-associated nanoparticles were analyzed by a Fluoroskan Ascent reader (Thermo Labsystems, Finland) using λex = 485 nm, λem = 530 nm. The percentage of uptake was expressed as the fluorescence intensity associated with cells versus the amount of fluorescence intensity present in the feed solution.Blood clearance and biodistribution in vivo
Blood clearance and biodistribution studies were performed using female ICR mice (25 ± 5 g). All animal experiments were in compliance with guidelines of the National Act on the use of experimental animal (People's Republic of China). The mice were fasted overnight but allowed free access to water. The mice, three per group, were injected intravenously into the tail vein with the various nanoparticle suspensions of 10 mg mL−1 at a nanoparticle suspension dose of 10 mL kg−1 (body weight of mice). After i.v. injection at designated time points (5 min, 15 min, 30 min, 1 h, 2 h, 3 h, 6 h, 12 h, 24 h, 36 h, 48 h), blood samples were collected from fossa orbitalis. Subsequently, the mice were sacrificed and the major organs (heart, liver, spleen, lung, kidney and brain) were excised and rinsed by Millipore water.Blood clearance and biodistribution of nanoparticles were determined by measuring the fluorescence of coumarin-6 after their extraction of nanoparticles from blood or organs into acetonitrile at λex = 485 nm and λem = 530 nm. The extraction procedure in detail was described by our group previously.23 The survival amount in blood then can be calculated. A logarithmic regression analysis allowed us to calculate the half-life (t1/2).
Statistical analysis
Data were expressed as mean ± SD. All data were generated in three or four independent experiments. Statistical analysis was performed using one-way analysis of variance (ANOVA). Differences were considered to be significant at a level of P < 0.05.Results and discussion
Characterization of the formulated LPHNPs
The physicochemical properties of various nanoparticles, including particle size, polydispersity index (PI), zeta potential, contact angle and EE (encapsulation efficiency) are listed in Table 1. As previously mentioned, the desired diameter of long-circulating colloidal carriers is within a relatively narrow range (70–200 nm), we firstly tailored the particle size in this size range. It can be found that the surface coating had only a small effect on the particle size distribution of nanoparticles. The average particle size of all the formulated LPHNPs ranged from 100 to 200 nm with a narrow size distribution (PI < 0.2).| Formulation | Average size (nm) | Polydispersity index | Zeta potential (mV) | Contact angle (°) | EE (%) |
|---|---|---|---|---|---|
| a All values indicate mean ± SD for n = 3 independent experiments. | |||||
| LPHNPs | 142.4 ± 10.2 | 0.151 ± 0.012 | 32.6 ± 1.2 | 97.46 ± 0.86 | 71.6 ± 1.1 |
| PEG LPHNPs | 128.1 ± 11.7 | 0.134 ± 0.008 | 24.7 ± 0.8 | 61.52 ± 0.73 | 77.2 ± 0.7 |
| HEP LPHNPs | 163.5 ± 11.3 | 0.148 ± 0.010 | −12.9 ± 1.3 | 34.61 ± 1.14 | 80.4 ± 1.0 |
| PEG/HEP LPHNPs | 131.5 ± 9.6 | 0.126 ± 0.009 | 5.7 ± 1.0 | 42.27 ± 0.85 | 86.7 ± 0.8 |
Zeta potential of the nanoparticles was found to be strongly influenced by the different surface coatings. LPHNPs possessed a high zeta potential value of 32.6 ± 1.2 mV. From this result, we can speculate that the cationic lipids covered the entire surface of the negative PLA shell. The zeta potential value of PEG LPHNPs was 24.7 ± 0.8 mV, which, substituting HEP for PEG, was inverted to negative value (−12.9 ± 1.3 mV). It can be noted that PEG/HEP LPHNPs exhibited an approximately neutral charge of 5.7 ± 1.0 mV. This diversity in zeta potential of the system is attributed to non-ionic nature of PEG and anionic nature of HEP, indicating the presence of PEG, HEP or PEG/HEP coating surrounding the LPHNPs.
The contact angle of bare LPHNPs is 97.46 ± 0.86, indicating a high hydrophobic nature. By contrast, a high improvement in surface hydrophilicity was observed for LPHNPs modified with PEG, HEP or PEG/HEP as evidenced by their significantly decreased static contact angles, 61.52 ± 0.73, 34.61 ± 1.14 and 42.27 ± 0.85, respectively.
LPHNPs yielded a DOX loading capacity (EE) of 71.6 ± 1.1%. After introducing PEG and HEP on the surface of LPHNPs, the EE increased by about 8% and 12%, respectively. The highest EE of 86.7 ± 0.8% was obtained in the case of PEG/HEP LPHNPs. It can be seen that the surface coating had a positive effect on the EE of nanoparticles, which helped to prevent the encapsulated drug from freely diffusing out of the nanoparticles during the solvent diffusion and evaporation process.
The typical morphology of PEG/HEP LPHNPs in freeze-dried form was shown in Fig. 1. It was obvious that the nanoparticles were in spherical shape with a smooth surface.
 | ||
| Fig. 1 SEM images of PEG/HEP LPHNPs. | ||
In vitro drug release profiles
Fig. 2 shows the profiles of DOX release from different formulations in the PBS at 37 °C. The results show that the release rate of DOX from the nanoparticles was in the order of LPHNPs > PEG LPHNPs > HEP LPHNPs > PEG/HEP LPHNPs. And, it can be seen that the release of DOX through these nanoparticles was rather slow. Especially, less than 10% DOX were released from the PEG/HEP LPHNPs after the 120 h period. Based on these in vitro data, it can be inferred that the PEG/HEP coating may prevent the DOX from leaking out of these nanoparticles during the prolonged circulation in vivo. | ||
| Fig. 2 Release profiles of DOX from LPHNPs or decorated with PEG, HEP or PEG/HEP incubated in PBS at 37 °C. Data represent mean ± SD, n = 3. | ||
Suspension stability of the LPHNPs
It is well-known that nano-sized particles are prone to aggregate in media, and the particle surface composition plays an important role in their stability. In this work, we have investigated the effect of PEG, HEP or PEG/HEP coating on the stability of nanoparticles in the presence of blood serum. The ΔBS as a function of time for LPHNPs with different surface coatings (PEG, HEP or PEG/HEP) were exhibited in Fig. 3. LPHNPs were also included for comparison. The variation of ΔBS at 120 h for control LPHNPs was 6.06%, indicating the lowest stability. By comparison, both PEG and HEP coatings had a positive effect at improving the stability of nanoparticles. Notably, despite having a surface charge close to neutrality, PEG/HEP LPHNPs exhibited the highest stability with ΔBS of 2.82%. We also determined the surface potentials of these nanoparticles during the variation. The results showed that the surface potentials of nanoparticles had no variation (almost the same as in Table 1) as the backscattering changed, which could not interfere with the ΔBS. | ||
| Fig. 3 Variation of ΔBS of LPHNPs or decorated with PEG, HEP or PEG/HEP in the presence of blood serum as a function of time (0–120 h). Data represent mean ± SD, n = 3. | ||
Previous studies showed the nanoemulsion with ionic surfactants had good stability when its zeta potential was above 30 mV.24,25 In our study, interestingly, PEG/HEP LPHNPs show high stability with a surface charge close to neutrality. One potential reason is the synergistic effect of hydrophilic PEG and HEP chains creating a shield between the nanoparticle surface and the surrounding medium, thereby inhibiting coalescence or flocculation.
Cytotoxicity of the LPHNPs
In this study, EVC-304 cells (endothelial cell line) were selected to better mimic the blood vessel wall where LPHNPs could potentially cause cytotoxicity in long-circulation.26 Fig. 4 shows the viability of cells incubated with LPHNPs coated with PEG, HEP and PEG/HEP with graded concentrations (from 0.0625 mg mL−1 to 1 mg mL−1) for a period of 48 h. The cytotoxicity of the various LPHNPs was concentration-dependent and the cell viability decreased slightly with the increasing concentration. We did not observe a significant difference (P > 0.05) in the toxicity of different formulations at any of the concentrations used. Although there was a slight reduction in the cell viability of LPHNPs covered by HEP alone, the combination of PEG with HEP could help in improving their biocompatibility. Average cell viabilities were between 77 and 115% of control at the concentrations studied, demonstrating that there was no significant cytotoxicity observed for LPHNPs varied in surface properties (PEG, HEP or PEG/HEP).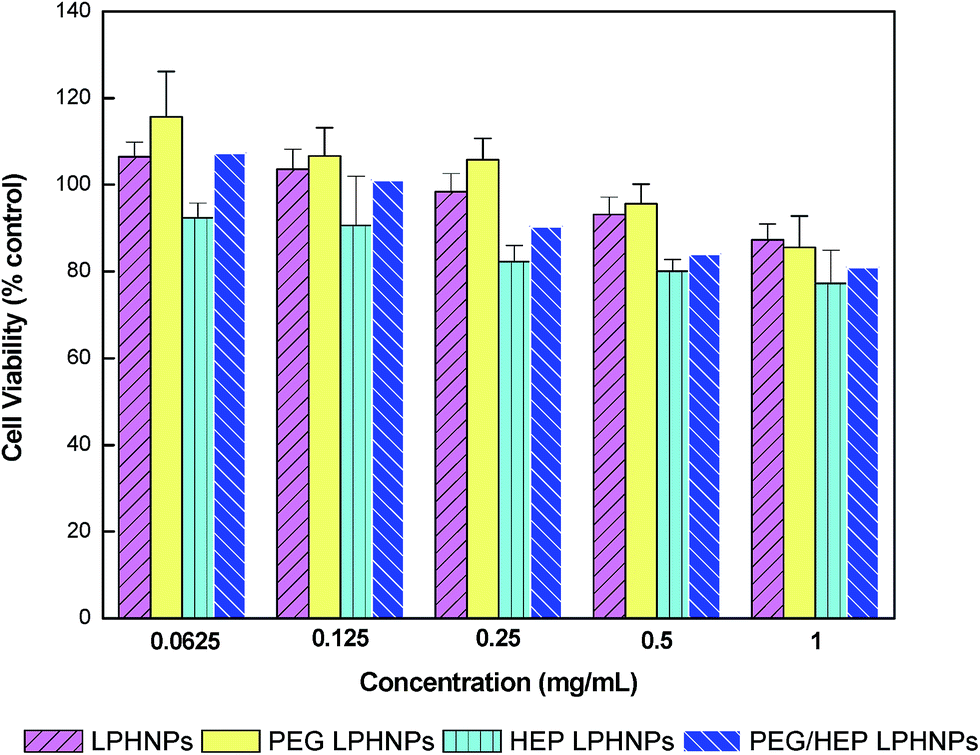 | ||
| Fig. 4 Cytotoxicity of LPHNPs or with various coatings (PEG, HEP, PEG/HEP) analyzed by MTT assay. ECV-304 cells were cultured with 0, 0.0625, 0.125, 0.25, 0.5, 1 mg mL−1 of LPHNPs for 48 h at 37 °C. Data represent mean ± SD, n = 4. | ||
In vitro phagocytic uptake
Coumarin-6, which has been extensively used as a fluorescent marker of polymer-base particles,9,27 was adopted as the fluorescent marker of LPHNPs to investigate their biological behaviors in this work. To validate the reliability of the data obtained from the in vitro and in vivo experiments, in vitro coumarin-6 release assay was carried out in advance (data not shown). The results revealed that the total released amount of coumarin-6 from the different formulations were less than 2% during the 72 h incubation period, indicating that the coumarin-6 labeled in the LPHNPs could not interfere with their inherent performance in vitro and in vivo.MPMs with phagocytic ability were chosen to mimic the in vivo conditions. Phagocytic uptake of NPs by MPMs is thought to be responsible for the major loss of nanoparticles in the systemic circulation. In this work, prior to the in vivo experiment, the effect of surface properties on the phagocytic uptake of coumarin-6 labeled LPHNPs in vitro was evaluated qualitatively and quantitatively, respectively. The percentage of cell-associated fluorescence intensities will be the definition for the uptake percentage. Fig. 5 exhibits the uptake efficiency of nanoparticles with different surface coatings (PEG, HEP or PEG/HEP) by MPM after 1 h incubation at 37 °C. The images of phagocytic uptake were shown in Fig. 5(a–d), and the statistical results were given in Fig. 5(e). It can be seen that the fluorescent intensities of various LPHNPs were in the order of LPHNPs > HEP LPHNPs > PEG LPHNPs > PEG/HEP LPHNPs. The uptake percentage of unmodified LPHNPs, PEG LPHNPs and HEP LPHNPs was 26.83%, 15.39% and 18.09%, respectively. The results demonstrated that, the presence of PEG or HEP coating alone had a slight effect on the reduction of phagocytic uptake. Interestingly, combining PEG with HEP clearly attenuated the nanoparticle–cell association, representing only 3.27% of cell uptake. The capture of PEG/HEP LPHNPs by MPM was 4.7-fold/5.5-fold lower than that of the corresponding PEG LPHNPs and HEP LPHNPs, respectively.
 | ||
| Fig. 5 (a–d) Microphotographs and (e) quantitative assay of phagocytosis of various nanoparticle formulations, which were measured after 1 h incubation with MPM in the serum containing medium at 37 °C. Data represent mean ± SD, n = 4. (a) LPHNPs, (b) PEG LPHNPs, (c) HEP LPHNPs, (d) PEG/HEP LPHNPs. Scale bar: 10 μm. | ||
Blood clearance and biodistribution in vivo
The in vivo blood clearance and biodistribution of the various LPHNPs were investigated to fully describe the in vivo fate of foreign nanoparticles. The blood concentration–time profiles after injection of various formulations (LPHNPs, PEG LPHNPs, HEP LPHNPs and PEG/HEP LPHNPs) in mice are illustrated in Fig. 6. The half-lives of the different LPHNPs calculated are shown in Fig. 7. Obviously, control LPHNPs were quickly removed from the systemic circulation with the shortest t1/2 of 0.3 h. Also, LPHNPs coated with PEG or HEP alone exhibited a rapid elimination from the bloodstream and the remaining dose at 6 h post-injection was 39.69% and 31.56%, respectively. Notably, LPHNPs modified with PEG and HEP achieved significant prolongation in blood circulation. There were still 55.26% of the fluorescence-associated PEG/HEP LPHNPs remaining in the systemic circulation at 48 h after injection. The t1/2 calculated for PEG/HEP LPHNPs was 72.6 h, 26.5-fold as long as that of PEG LPHNPs and 48.7-fold as long as that of HEP LPHNPs. Furthermore, though all types of LPHNPs show biphasic clearance profile, an obvious retardation in clearance from the blood was observed in PEG/HEP LPHNPs group in the initial 6 h after injection. | ||
| Fig. 6 Blood clearance profile of surface-modified nanoparticle formulations (LPHNPs, PEG LPHNPs, HEP LPHNPs and PEG/HEP LPHNPs) after i.v. injection in mice. Data represent mean ± SD, n = 3. | ||
 | ||
| Fig. 7 Half-life (t1/2) in blood circulation of various nanoparticle formulations (LPHNPs, PEG LPHNPs, HEP LPHNPs and PEG/HEP LPHNPs). Data represent mean ± SD, n = 3. | ||
Interestingly, surface modification of nanoparticles with PEG or HEP alone failed to reduce the macrophage uptake as well as prolong the circulatory half-life. However, after combining PEG with HEP, a significant phagocytic uptake inhibition and blood circulation prolongation was observed. The possible reasons are analyzed as follows. First, previous studies demonstrated that the stronger the surface hydrophobicity, the more easily it would be adsorbed by plasma proteins and thus recognized by the RES.28,29 As the PEG/HEP coating exhibited more hydrophilicity than the other coatings, it may possibly adsorb more water molecules around the particle surface to form a ‘cloud’ of hydration layer in the aqueous environment, which might effectively avoid the hydrophobic interactions with plasma proteins, protecting them from recognizing by phagocytes. Second, it has been reported that neutrally charged particles have a much lower opsonization than positively or negatively charged particles.30 The surface modification of LPHNPs using the combined neutral PEG and negative HEP synergistically tailored the positive hybrid particle surface close to neutrality, which inhibited the electrostatic interactions with opsonins. Their lack of binding with opsonins and then evading phagocytosis would be likely responsible for their prolonged persistence in the blood circulation. By contrast, PEG or HEP coating alone failed to neutralize the surface charge of LPHNPs. Third, it was supposed to be resulted from the surface molecular conformation. It has been hypothesized that the surface-bound PEG chains would shape into either a ‘mushroom’ or a ‘brush’ conformation depending on their surface density, and the latter conformation would create more effective blocking or repulsion of opsonins than the former one.31,32 We can postulate that, HEP groups possessing an extremely strong electrostatic affinity may overcome the osmotic repulsions between PEG chains to anchor on the particle surface. Consequently, the steric occupation of HEP chains on the particle surface might facilitate more stretching of neighbored PEG chains. Thus, the surface-grafted PEG in combination with HEP was supposed to shape a dense brush-like conformation.
In this work, PEG/HEP LPHNPs exhibiting the most prolonged blood circulating property was chosen to investigate the in vivo fate of nanoparticles after i.v. administration. The distributions of PEG/HEP LPHNPs and control LPHNPs in various organs (heart, liver, spleen, lung and kidney) for a 48 h period after administration are illustrated in Fig. 8. It can be seen that, the nanoparticles were predominantly found in the liver, followed by the kidney and spleen, to a lesser extent, in the lung and heart. In the case of PEG/HEP LPHNPs, the cumulative amount compared with that of LPHNPs dramatically reduced in liver, changed little in other organs. This reduction in uptake of PEG/HEP LPHNPs by liver macrophages (Kupffer cells) correlated well with a noted retention of circulatory level in the blood at the initial period. It is well-known that the nanoparticles cleared from the blood were mainly captured by MPS organs. Since liver is the main MPS organ, there seems logical to exist a correlation between the level of increased blood retention and their reduced sequestration by liver. Within the liver, they displayed a fast accumulation in the first 5 min followed by hepatobiliary excretion out to 48 h. The rate of accumulation in liver for PEG/HEP LPHNPs was 3-fold lower than that of LPHNPs at the initial 5 min. This significant reduction in liver was probably due to the ‘invisible’ surface created by the hydrophilicity PEG/HEP coating with neutral-charge and brush-like conformation, which effectively avoids recognition by liver macrophages and subsequent hepatic deposition.
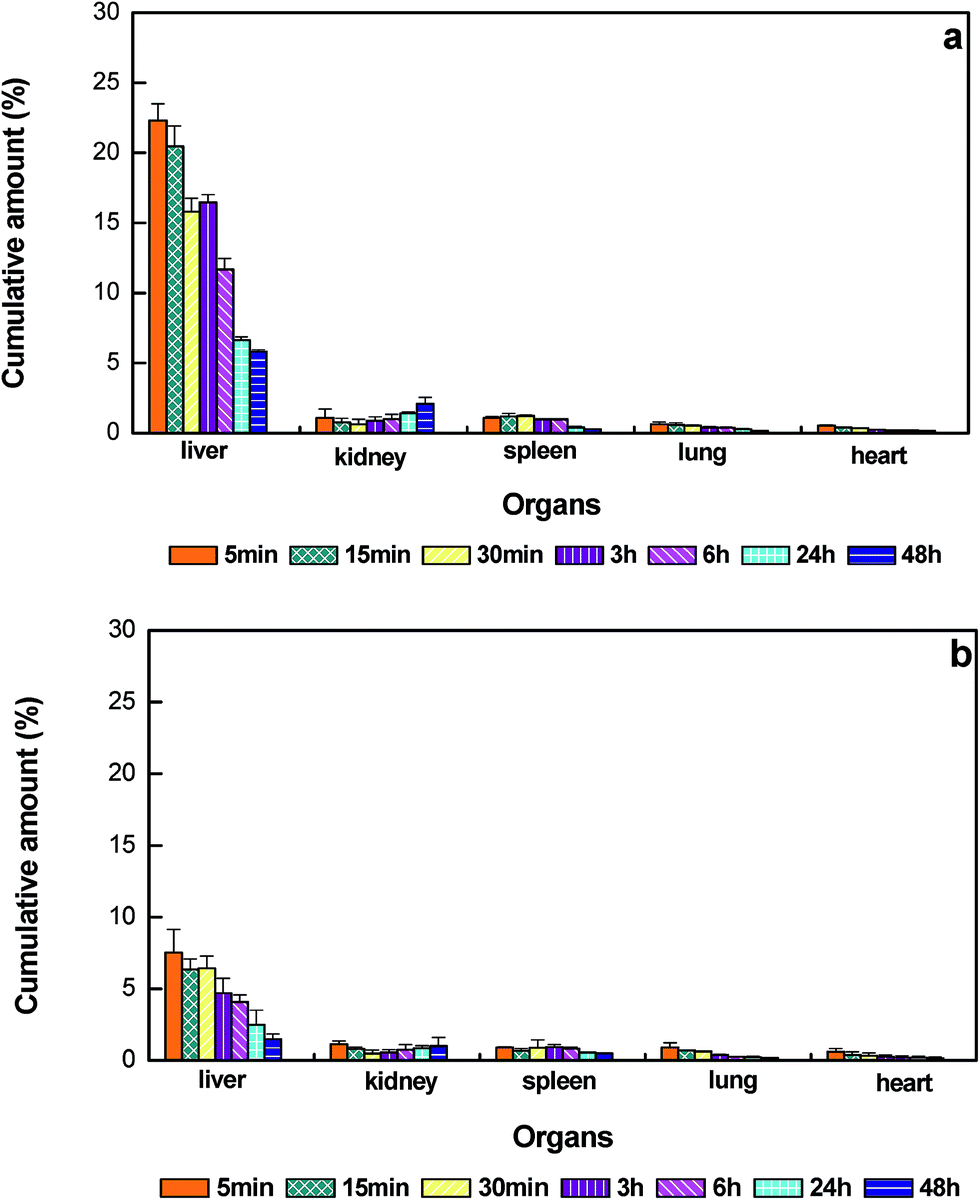 | ||
| Fig. 8 The cumulative percentage of nanoparticles in main organs over time after i.v. injection in mice. (a) LPHNPs, (b) PEG/HEP LPHNPs. Data represent mean ± SD, n = 3. | ||
Currently, there is no a clear standard to evaluate if the half-life of the reported nanocarrier in blood circulation is enough for the efficient tumor targeted drug delivery. However, clearly, nanoparticles showing improved suspension stability and long-circulation in vivo could have more opportunities for systemic circulation into different tumor sites in patients, as compared counterparts. Based on our experiments with PEG/HEP LPHNPs, an active tumor targeting drug carrier can be designed through bio-conjugation of affinity ligands, such as antibodies, peptides, sugars and small molecules to the surface of PEG/HEP LPHNPs without impairing their long-circulating properties. Theoretically, they are expected to achieve targeted delivery by specifically binding to antigens overexpressed on the tumor cells. Further study will focus on the in vivo drug delivery based on these long-circulating PEG/HEP LPHNPs for tumor targeting and treatment.
Conclusions
In this study, we developed LPHNPs bearing a combinatorial coating of PEG and HEP for the application of long-circulating drug delivery. The PEG/HEP LPHNPs show homogeneous particle size, high surface hydrophilicity and neutral surface charge. The cytotoxicity results indicated the LPHNPs with PEG/HEP coating were non-toxic for EVC-304 cells and their in vitro phagocytic uptake by MPM was greatly reduced. In vivo studies in ICR mice showed that the blood circulation half-life of PEG/HEP LPHNPs was 234-fold than that of control LPHNPs, with reduced amount sequestered by the liver. These results demonstrated that these stealth LPHNPs with PEG/HEP coating may have a great promise as effective drug carriers that can suffice for good long-circulating efficacy to reach its target.Acknowledgements
The authors wish to express their gratitude to the financial supports from National Natural Science Foundation of China (No. 31300789) and Natural Science Foundation of Shandong Province (No. ZR2013CQ016).References
- X. Z. Yang, S. Dou, Y. C. Wang, H. Y. Long, M. H. Xiong, C. Q. Mao, Y. D. Yao and J. Wang, ACS Nano, 2012, 6, 4955–4965 CrossRef CAS PubMed.
- X. Zhao, F. Li, Y. Li, H. Wang, H. Ren, J. Chen, G. Nie and J. Hao, Biomaterials, 2015, 46, 13–25 CrossRef CAS PubMed.
- N. Pippa, E. Kaditi, S. Pispas and C. Demetzos, J. Nanopart. Res., 2013, 15, 1685–1700 CrossRef.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trubetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600–1603 CrossRef CAS PubMed.
- A. S. Zahr, C. A. Davis and M. V. Pishko, Langmuir, 2006, 22, 8178–8185 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS PubMed.
- C. Fang, B. Shi, Y. Y. Pei, M. H. Hong, J. Wu and H. Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27–36 CrossRef CAS PubMed.
- J. Zhao, Y. D. Chai, J. Zhang, P. F. Huang, K. Nakashima and Y. K. Gong, Acta Biomater., 2015, 16, 94–102 CrossRef CAS PubMed.
- S. Parveen and S. K. Sahoo, Eur. J. Pharmacol., 2011, 670, 372–383 CrossRef CAS PubMed.
- Y. Sheng, Y. Yuan, C. S. Liu, X. Y. Tao, X. Q. Shan and F. Xu, J. Mater. Sci.: Mater. Med., 2009, 20, 1881–1891 CrossRef CAS PubMed.
- H. J. Gao, J. J. Liu, C. H. Yang, T. J. Cheng, L. P. Chu, H. Y. Xu, A. M. Meng, S. J. Fan, L. Q. Shi and J. F. Liu, Int. J. Nanomed., 2013, 8, 4229–4246 Search PubMed.
- B. G. Li, Q. Wang, X. Wang, C. Z. Wang and X. Q. Jiang, Carbohydr. Polym., 2013, 93, 430–437 CrossRef CAS PubMed.
- J. Zhang, M. C. Shin, A. E. David, J. Zhou, K. Lee, H. N. He and V. C. Yang, Mol. Pharmaceutics, 2013, 10, 3892–3902 CrossRef CAS PubMed.
- Y. Sheng, C. S. Liu, Y. Yuan, X. Y. Tao, F. Yang, X. Q. Shan, H. J. Zhou and F. Xu, Biomaterials, 2009, 30, 2340–2348 CrossRef CAS PubMed.
- R. A. H. Ishak, G. A. S. Awad, N. M. Zaki, A. E. H. A. El-Shamy and N. D. Mortada, Carbohydr. Polym., 2013, 94, 669–676 CrossRef CAS PubMed.
- Y. P. Li, Y. Y. Pei, X. Y. Zhang, Z. H. Gu, Z. H. Zhou, W. F. Yuan, J. J. Zhou, J. H. Zhu and X. J. Gao, J. Controlled Release, 2001, 71, 203–211 CrossRef CAS PubMed.
- N. M. Khalil, T. C. F. do Nascimento, D. M. Casa, L. F. Dalmolin, A. C. de Mattos, I. Hoss, M. A. Romano and R. M. Mainardes, Colloids Surf., B, 2013, 101, 353–360 CrossRef CAS PubMed.
- Y. Sheng, C. S. Liu, Y. Yuan, X. L. Zhang, X. Q. Shan and F. Xu, J. Biomed. Mater. Res., Part B, 2009, 91B, 631–642 CrossRef CAS PubMed.
- S. S. Cao, B. L. Liu, X. B. Deng, R. Luo and H. L. Chen, Polym. Int., 2007, 56, 357–363 CrossRef CAS.
- V. M. Gaspar, A. F. Moreira, E. C. Costa, J. A. Queiroz, F. Sousa, C. Pichon and I. J. Correi, Colloids Surf., B, 2015, 134, 287–294 CrossRef CAS PubMed.
- Y. Sheng, H. J. He and H. Zou, Drug Delivery, 2014, 21, 370–378 CrossRef CAS PubMed.
- J. Zhao, C. S. Liu, Y. Yuan, X. Y. Tao, X. Q. Shan, Y. Sheng and F. Wu, Biomaterials, 2007, 28, 1414–1422 CrossRef CAS PubMed.
- M. Bivas-Benita, S. Romeijn, H. E. Junginger and G. Borchard, Eur. J. Pharm. Biopharm., 2004, 58, 1–6 CrossRef CAS PubMed.
- J. K. Jiang, G. Oberdorster and P. Biswas, J. Nanopart. Res., 2009, 11, 77–89 CrossRef CAS.
- T. Liu, Y. Liu, Y. Chen, S. H. Liu, M. F. Maitz, X. Wang, K. Zhang, J. Wang, Y. Wang, J. Y. Chen and N. Huang, Acta Biomater., 2014, 10, 1940–1954 CrossRef CAS PubMed.
- K. Y. Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS PubMed.
- M. Luck, B. R. Paulke, W. Schroder, T. Blunk and R. H. Muller, J. Biomed. Mater. Res., 1998, 39, 478–485 CrossRef CAS PubMed.
- J. J. Luan, X. Y. Yang, L. J. Chu, Y. W. Xi and G. X. Zhai, J. Colloid Interface Sci., 2014, 428, 49–56 CrossRef CAS PubMed.
- M. Roser, D. Fischer and T. Kissel, Eur. J. Pharm. Biopharm., 1998, 46, 255–263 CrossRef CAS PubMed.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Langmuir, 2005, 21, 12327–12332 CrossRef CAS PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier and J. P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |