
Computational insight into conformational states of glucagon-like peptide-1 receptor (GLP-1R) and its binding mode with GLP-1
Juan Zhanga,
Shikai Gua,
Xianqiang Sunb,
Weihua Lia,
Yun Tang*a and
Guixia Liu*a
aShanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China. E-mail: gxliu@ecust.edu.cn; ytang234@ecust.edu.cn; Fax: +86-21-64253651; Tel: +86-21-64250811
bDepartment Division of Theoretical Chemistry and Biology, School of Biotechnology, KTH Royal Institute of Technology, S-106 91 Stockholm, Sweden
First published on 27th January 2016
Abstract
The glucagon-like peptide-1 receptor (GLP-1R) has captivated researchers because of its tremendous therapeutic effects for the treatment of type 2 diabetes mellitus (T2DM). However, since the full-length crystal structure of GLP-1R has not been revealed yet, the molecular binding mode and the activation mechanism remain unclear, which will be the obstacle for the discovery of novel potent GLP-1R agonists. In the present study, we constructed the model of GLP-1R in its full length and explored the binding modes between GLP-1 and GLP-1R by means of a bunch of computational methods including homology modeling, protein–protein docking, and molecular dynamics simulations. Our model is in agreement with previous experiment and the results from our MD simulations that verified the binding modes between GLP-1 and GLP-1R are reasonable. What's more, we found the absence or presence of GLP-1 significantly affected the conformation of extracellular domain (ECD) of GLP-1R. The GLP-1R in the apo form stabilized in a ‘closed’ state which is unfavorable to the binding of GLP-1, resembling as the GCGR. By contrast, in the GLP-1/GLP-1R complex, GLP-1R maintained an ‘open’ state.
1. Introduction
The glucagon-like peptide-1 receptor (GLP-1R) belongs to the secretin like (class B) family of G-protein-coupled receptors (GPCRs).1–3 Exactly the same as all the other class B GPCRs, GLP-1R contains a seven-transmembrane domain (TMD) and a relatively long N-terminal extracellular domain (ECD). Glucagon-like peptide-1 (GLP-1) is the endogenous agonist of GLP-1R and secreted from the gastrointestinal tract in response to food intake.4 The ECD constitutes a large region for the binding of GLP-1 to GLP-1R. Upon the binding of GLP-1 to GLP-1R, the receptor is activated and the insulin secretion is enhanced in a glucose-dependent manner.5,6 In addition, such binding reduces food intake by inhibiting gastric acid secretion and slows down gastric empty, possibly through a central effect, and causes weight loss. Therefore, GLP-1R has attracted significant attention for the treatment of type 2 diabetes mellitus (T2DM) and obesity.4 GLP-1 therapy was reported to exhibit potential cardioprotective effects as well.7GLP-1 is comprised of 30 amino acids8 and GLP-1 mimicking drugs have been extensively studied because of its medicinal value. Currently, a couple of GLP-1 mimicking drugs9–14 have been approved or in clinical trials such as exenatide15–17 and liraglutide.18,19 Prior to other diabetes drugs, GLP-1 mimicking drugs reduce the risk of hypoglycemia when used alone or together with metformin.20
The resolvation of crystal structures of class B GPCR, TMDs of GCGR21 and CRF1R,22,23 greatly advanced the studies of GLP-1R by providing a reliable homology for the homology modeling of the structure of GLP-1R. It is interesting that the ECD domain of GLP-1R coupled with GLP-1 has been determined24 (PDB ID 3IOL), and thus provides detailed interactions between the GLP-1 and the ECD domain. However, a complete crystal structure for the GLP-1R or GLP-1/GLP-1R complex remains unresolved, which hampers our understanding of the binding mode and the activation mechanism of GLP-1/GLP-1R.
To address this issue, our strategy was combining homology modeling, protein–protein docking, and molecular dynamics simulations to understand the binding mode of GLP-1 to GLP-1R. The GLP-1/GLP-1R complex structure agrees well with published experimental studies.25,26 We suggest that the extracellular loop 3 (ECL3) of GLP-1R plays important role in ligand binding and receptor activation. Once the structure of GLP-1/GLP-1R complex or GLP-1R in the apo state was subjected to the MD simulation, we observed that the apo-GLP-1R stabilized in a ‘closed’ state resembling as GCGR, while GLP-1R coupled with GLP-1 in the ‘open’ state. Our study thus provides atomic information for drug designing targeting GLP-1R and also sheds light for the activation mechanism of GLP-1R.
2. Methods
2.1 Construction of complete GLP-1/GLP-1R complex model
2.2 Structure validation and energy minimization
After the complete models of GLP-1/GLP-1R were produced, MacroModel 9.9 in Schrödinger software package was employed to perform energy minimization calculations for the final model: first, fixed the whole structure except the hydrogen atom and optimized the hydrogen atom, and then fixed the backbone of complex structure and optimized the side chain atoms, and finally removed any constraint and optimized the whole structure. And the parameters defined in MacroModel were set as follows: OPLS_2005 was selected as force field; solvation treatment was set to octanol, and constraints were set for atoms. Furthermore, the qualities of these models were evaluated by Profile-3D program. Besides, we referred to previous publications25,26,28–30 and residue mutation study relevant to the interaction between GLP-1 and GLP-1R when comparing these models. Considering both verify score of Profile-3D and previous work, the most reasonable model was finally selected for further refinement. PROCHECK Ramachandran plot was also employed to validate model stereochemical quality.2.3 System setup and molecular dynamics simulations
A complete GLP-1/GLP-1R complex model was constructed through aforementioned steps. While GLP-1R model in apo-form was obtained by removing the GLP-1 from the GLP-1/GLP-1R complex model (namely apo-GLP-1R). After two systems were set up, long-time molecular dynamics (MD) simulations were carried out.In order to be closer to the real environment in human body, two systems were embedded into a pre-equilibrated POPC lipid bilayer with the surface area of 75 Å × 75 Å on the X–Y plane using in-house program, respectively. Membrane orientation was determined according to the orientations of proteins in membranes (OPM) database. Two protein-POPC systems were solvated by a 75 Å × 75 Å × 140 Å box full of water molecules. POPC molecules within 0.8 Å of the models and water molecules in the bilayer were removed. The same number of chloride ions and sodium ions were added to two neutral systems with a concentration of 0.15 M. The constitution of two molecular dynamics systems was listed in Table 1. The detailed procedure for MD simulations was adopted referring to our previous studies.31,32
| System | POPC | Na+ | Cl− | H2O |
|---|---|---|---|---|
| apo-GLP-1R | 105 | 71 | 71 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 906 906 |
| GLP-1/GLP-1R | 105 | 71 | 71 | 17800 |
All MD simulations were performed using Gromacs 4.6.5 (ref. 33 and 34) package in a periodic boundary condition. The CHARMM 36 force field fit for the proteins, lipids, and ions was applied and TIP3P model was employed as the water model. Prior to MD simulations, each system was subject to 50000-step energy minimization to relieve internal repulsions with the steepest descent algorithm and a maximum force of 10.0 kJ mol−1 nm−1. After energy minimization, NVT equilibration was conducted for 50 ps at 310 K using the Nose–Hoover for temperature control. After stabilizing the temperature, NPT equilibration was performed for another 1 ns at 1 bar using the Parrinello–Rahman pressure coupling. NVT and NPT equilibrations were run successively with position restraints on the GLP-1R/POPC or GLP-1/GLP-1R/POPC. The long-range electrostatics was set to the particle mesh Ewald algorithm, and constraints for all bonds were applied using the LINCS algorithm. Following NVT and NPT equilibration, 220 ns production MD was carried out for two systems using an NPT ensemble with a time step of 2 fs. Production trajectories analysis was performed by GROMACS in-built analysis tools.
In order to calculate the average energy of the GLP-1R in two kind of states, 81 snapshots were extracted every 0.25 ns from 15–35 ns and 200–220 ns production trajectories respectively and the energy were calculated by MacroModel 9.9.
3. Results and discussion
3.1 Structure of GLP-1/GLP-1R complex
Up to now, unlike the explosion of crystal structure of GPCRs in class A, only two crystal structures of the transmembrane domain of GPCRs in class B, GCGR and CRF1R, were resolved. GCGR shares high homology with GLP-1R and both GCGR and GLP-1R belong to the ‘glucagon receptor family’.35 The corresponding ligands towards GLP-1R and GCGR are GLP-1 and glucagon, respectively, which are also highly conserved in sequence. Thus, the TMD crystal structure of GCGR was used to build the TMD structure of GLP-1R. The sequence alignment used in the homology modeling was shown in Fig. 1. The sequence identity between the two receptors is 54.0% and sequence similarity is 74.6%, which are superior to previous work.26,36 | ||
| Fig. 1 Sequence alignment between the transmembrane domain of target protein GLP-1R and template protein GCGR. Sequence identity is 54.0%, and sequence similarity is 74.6%. | ||
Biologically active GLP-1 consists of 30 or 31 amino acids and the main active form is 7-36NH2.37 It was reported that the first three residues (H1-E3) of GLP-1 (7-36)-NH2 play an important role in triggering GLP-1R activation.38 However, the sequencing of GLP-1 is only from G4 to G29 in the crystal structure of GLP-1 in complex with the extracellular domain of the GLP-1R (PDB ID 3IOL). To obtain the structure of GLP-1R with an active GLP-1, we replaced GLP-1 (G4-G29) in the crystal structure (PDB ID 3IOL) with GLP-1 (H1-R30) in the NMR structure (PDB ID 1D0R). Since the conformation of GLP-1 in the active form remains unknown, 20 various conformations of GLP-1 in the NMR structure were all retained for protein–protein docking. Then, we got complete GLP-1/GLP-1R complexes by docking the 20 GLP-1s bound to the ECD of GLP-1R to the TMD model of GLP-1R. In previous work,26,36 GLP-1 was manually docked to the transmembrane domain, and GLP-1/GLP-1R complex was manually assembled guided by the key interactions between GLP-1 and GLP-1R. To some extent, relative to manual operation, precise docking software ZDOCK behaves better in predicting binding conformation of GLP-1/GLP-1R complex.
Out of the GLP-1s in 20 conformations, three of them failed to be docked to the reasonable area of GLP-1R and were neglected in the following studies. The ECLs of GLP-1R in each of the 17 complex structures obtained from docking were further optimized to accommodate the GLP-1. Among the 17 complex structures, two of them can't be successfully optimized using PLOP and thus abandoned. Additionally, the loop linking the ECD and the TMD in other four complexes blocked the binding pocket and were discarded. Finally, a total of 11 GLP-1/GLP-1R complex models were produced.
The complex models were evaluated using a bunch of programs. All these models got high scores when evaluated using Profile-3D (Table 2). The Ramachandran plot of Model_20 (Fig. 2) showed that 99.7% of the residues were placed in allowed regions, and only 0.3% of the residues were in the disallowed regions. The evaluations by Profile-3D and Ramachandran plot indicated that Model_20 was in good quality and was reliable as starting structure of MD simulations. Considering the Profile-3D score of Model_20 was the second highest and it also agreed well with information from previous work, therefore, Model_20 was selected for the following MD simulations.
| Model ID | Verify score | Expected high score | Expected low score |
|---|---|---|---|
| Model_2 | 101.00 | 197.15 | 88.72 |
| Model_3 | 98.41 | 197.15 | 88.72 |
| Model_6 | 99.57 | 197.15 | 88.72 |
| Model_7 | 100.6 | 197.15 | 88.72 |
| Model_9 | 101.55 | 197.15 | 88.72 |
| Model_12 | 87.90 | 197.15 | 88.72 |
| Model_14 | 92.91 | 197.15 | 88.72 |
| Model_15 | 103.06 | 197.15 | 88.72 |
| Model_17 | 98.02 | 197.15 | 88.72 |
| Model_18 | 101.27 | 197.15 | 88.72 |
| Model_20 | 101.92 | 197.15 | 88.72 |
 | ||
| Fig. 2 The Ramachandran plot for analyzing the rationality of Model_20 structure. Residues in most favoured regions: 85.3%; residues in additional allowed regions: 11.9%; residues in generously allowed regions: 2.5%; residues in disallowed regions: 0.3%. | ||
3.2 Binding modes of GLP-1/GLP-1R optimized by MD simulations
A complete crystal structure for the GLP-1/GLP-1R complex remains unresolved, which hinders further understanding of the structure and function of GLP-1R. It's worthwhile to explore different states of GLP-1R and analyze the interaction between GLP-1 and GLP-1R relevant to the activation mechanism. Models of GLP-1/GLP-1R complex and apo-GLP-1R were obtained as described in the method section. Then, long-time MD simulations for GLP-1/GLP-1R complex and apo-GLP-1R systems were carried out aiming at exploring their dynamic characteristics. In two cases, two systems appeared to reach a stable state in the last 20 ns, as indicated by the RMSD values slightly fluctuated around a certain value (Fig. 3(a)). We analyzed the binding mode between GLP-1 and GLP-1R optimized by MD simulations in detail. | ||
| Fig. 3 (a) The RMSD values along the time frame for all atoms of the holo-GLP-1R (the GLP-1R in GLP-1/GLP-1R complex, red line) and the apo-GLP-1R (black line); (b) the RMSD values along the time frame for the backbone atoms of the ECD and TMD of holo-GLP-1R (blue line for ECD, green line for TMD), apo-GLP-1R (magenta line for ECD, cyan line for TMD), respectively. | ||
For the GLP-1/GLP-1R complex system, it can be observed that the ECD was stabilized in a conformation nearly perpendicular to the membrane surface. Through a long-time scale MD simulation, GLP-1 formed stable and extensive contacts (Fig. 4) with GLP-1R to maintain its stabilized conformation. Some key residues, Y152, R190, E364, and R380 of GLP-1R and H1, A2, E3, G4, T5, F6, D9, V10, Y13 of GLP-1,25,26,39 that have been identified to contribute to ligand binding and receptor activation in mutation study can be validated in our GLP-1/GLP-1R complex model. Specifically, H1 forms hydrogen bond with R190 (Fig. 4(b)). The nitrogen atom of imidazole ring of H1 also forms hydrogen bond with the oxygen atom of backbone of V229, suggesting the important role of imidazole ring for GLP-1 action, and this is consistent with previous experiment.38 E3 of GLP-1 forms H-bond network with Y148 and Y152. G4 and T5 of GLP-1 form hydrogen bond with E364. It can be observed that R380 of GLP-1R is involved in interactions with D9 of GLP-1 including salt bridges and H-bonds. Mutation of either D9 or R380 causes huge loss of affinity and potency of GLP-1.25 R380 and D9 are both evolutionarily conserved residues in class B GPCRs and their corresponding peptide ligands, respectively. Additionally, mutagenesis studies have confirmed the existence of such similar ligand–receptor interaction in two other class B GPCRs: GCGR (D9:R378) and GCRPR (D9:R379). The occupancies of hydrogen bonds in 200–220 ns trajectories are shown in Table 3.
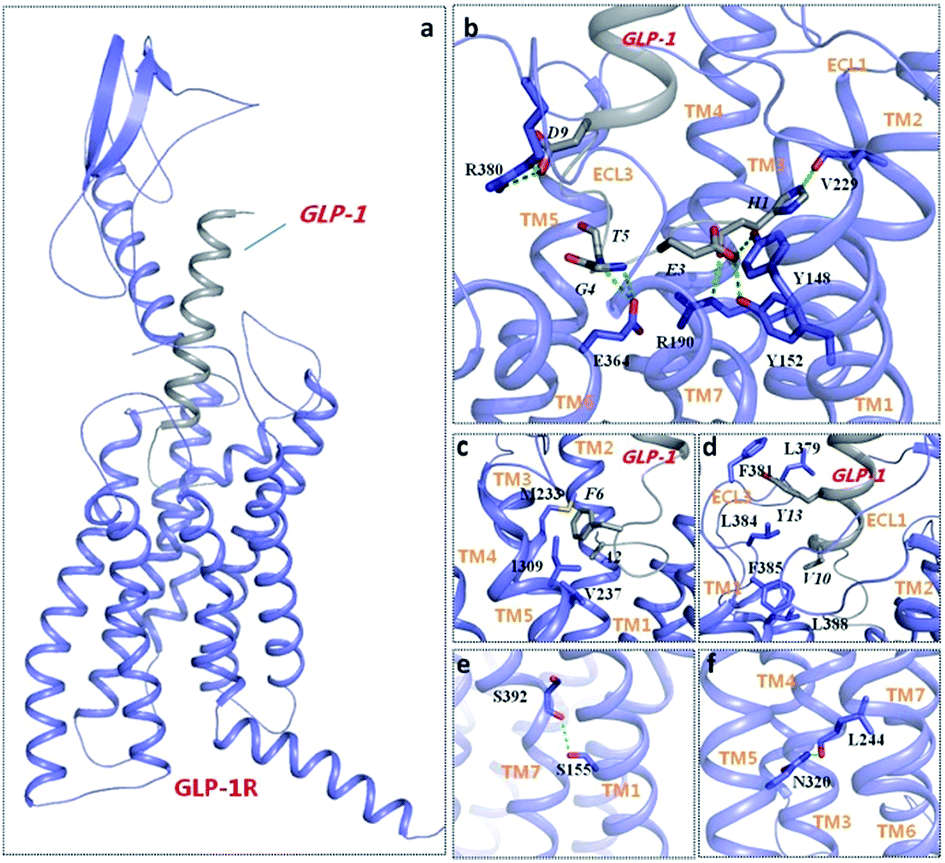 | ||
| Fig. 4 The typical structure of GLP-1/GLP-1R complex (a) in the MD simulations, polar (b) and hydrophobic (c and d) interactions between GLP-1 (grey) and GLP-1R (purple), and class B GPCRs conserved feature in the model (e and f). Residues involved in interactions between GLP-1 and GLP-1R are labeled with italics and regular font, respectively. Hydrogen bonds are depicted as green dotted lines. | ||
| GLP-1 | GLP-1R | Occupancy (%) |
|---|---|---|
| HIS1O | ARG190NE | 66.72 |
| HIS1NE2 | VAL229O | 87.91 |
| GLU3OE1 | TYR148OH | 87.96 |
| GLU3OE1 | TYR152OH | 74.71 |
| GLY4N | GLU364OE2 | 45.53 |
| THR5N | GLU364OE2 | 74.81 |
| ASP9OD1 | ARG380NE | 21.39 |
| ASP9OD2 | ARG380NE | 64.57 |
Besides polar interaction, F6 of GLP-1 orientates toward the top of TM3 and TM5 and locates in a hydrophobic pocket, forming favorable hydrophobic interactions with M233, V237 and I309 of GLP-1R. A2 of GLP-1 also shows hydrophobic interactions with V237 (Fig. 4(c)). In addition, Y13 resides in a hydrophobic cavity, forming hydrophobic interactions with L379, F381 and L384. V10 also forms hydrophobic interactions with L384, F385 and L388, which further stable ECL3 conformation of GLP-1R (Fig. 4(d)). More details about the conserved feature of class B GPCR can be found to prove the reasonability of our model: S155 (TM1) forms a H-bond with S392 (TM7), which is a conserved feature across class B GPCRs (Fig. 4(e)). Mutation of S155 and S392 of GLP-1R can alter receptor signaling.40 Another hydrogen bond is formed between N320 and L244 at the TM3-TM5 interface (Fig. 4(f)), which is a specific character among class B GPCR.
By aligning the typical snapshot in the simulation to the ECD crystal structure of GLP-1R coupled with GLP-1, it was observed that the C-terminal of GLP-1 can superimpose with each other very well, with two polar contacts and all the hydrophobic contacts are all retained (Fig. 5). In contrast, the N-terminal of GLP-1 deviates from the crystal structure. We attributed the conformational change of the N-terminal to the lacking of the first three residues (H1-E3) and the low active form of GLP-1 adopted in the crystal structure.
 | ||
| Fig. 5 Structure alignment of the typical snapshot of GLP-1/GLP-1R complex (GLP-1R is colored purple and GLP-1 is colored grey) in MD simulations with the ECD crystal structure of GLP-1R coupled with GLP-1 (PDB ID 3IOL; GLP-1R is colored cyan and GLP-1 is colored yellow). | ||
3.3 Open/closed conformational states of GLP-1R
It's obvious that the RMSD values of apo-GLP-1R increased during 35–50 ns from about 7 Å to 11 Å and was much higher than those of holo-GLP-1R (Fig. 3(a)). The apo-GLP-1R was supposed to undergo a large conformational change during 35–50 ns. To further figure out structural change that apo-GLP-1R went through, we calculated the temporal evolution of RMSD values of the ECD and TMD for apo-GLP-1R and holo-GLP-1R, respectively (Fig. 3(b)). It's interesting to find that RMSD values of the TMD were much lower and more stable than those of the ECD for both of apo-GLP-1R and holo-GLP-1R. Furthermore, the RMSD values of the ECD of apo-GLP-1R were higher than those of holo-GLP-1R. The RMSD values indicated that the ECD of apo-GLP-1R contributed the most to the overall conformational change. By comparison of apo-GLP-1R and holo-GLP-1R, we can get the conclusion that the absence of GLP-1 affected significantly the conformation of ECD of apo-GLP-1R. We clustered the 2000 snapshots in the last 20 ns of MD stimulations with a cutoff of 4 Å for both of apo-GLP-1R and holo-GLP-1R and analyzed GLP-1R structure in detail. The snapshots of apo-GLP-1R were classified into 6 clusters and the biggest cluster contained 1768 snapshots. And the snapshots of holo-GLP-1R were classified into 8 clusters and the biggest cluster contained 1266 snapshots.Obvious motions of the ECD domain towards the TMD can be found in the MD simulation of apo-GLP-1R. The ECD swung and rotated down toward the transmembrane domain, got closer to ECL3, reached a state of equilibrium. It can be observed that ECD contacts strongly with the third extracellular loop (ECL3) of GLP-1R, which stabilized the whole conformation of GLP-1R (Fig. 6). The residue S94 in ECD forms hydrogen bonds with E373 and H374. E128 forms stable contacts with R376 and R380 including salt bridge and hydrogen bond interactions. S135 also makes hydrogen-bond interaction with R376. These interactions allow ECD to contact ECL3 tightly, maintaining apo-GLP-1R in a stable state. The occupancies of hydrogen bonds in 200–220 ns trajectories are shown in Table 4.
 | ||
| Fig. 6 The typical structure of apo-GLP-1R in the MD simulations and interactions between the ECD and ECL3. Hydrogen bonds are depicted as green dotted lines. | ||
| ECD | TMD | Occupancy (%) |
|---|---|---|
| SER94OG | GLU373OE1 | 34.28 |
| SER94OG | HSE374N | 26.29 |
| GLU128OE1 | ARG376NH2 | 86.21 |
| GLU128OE1 | ARG380NH1 | 66.92 |
| GLU128OE2 | ARG376NH1 | 86.61 |
| GLU128OE2 | ARG380NH2 | 75.31 |
| SER135OG | ARG376NH2 | 70.66 |
The root-mean-square fluctuations (RMSF) of apo-GLP-1R and holo-GLP-1R are shown as (Fig. 7). Residues in the 3 ICLs and 3 ECLs showed large fluctuations as indicated in the Fig. 7. However, the third extracellular loop of apo-GLP-1R shows a relatively smaller RMSF value than that of GLP-1/GLP-1R complex. For apo-GLP-1R, interactions between the ECL3 and the ECD of GLP-1R stabilized ECL3, while ECL3 of the complex reflected a larger displacement because of the involvement of GLP-1. Interactions (R380:D9; L379, F380:Y13) between the ECL3 and GLP-1 highlighted the role of ECL3 in ligand binding and activation. The flexible N-terminal of GLP-1 allowed it to stretch deep into the cavity of GLP-1R formed by TM1, TM2, TM3, TM5, TM6, TM7, ECL2 and ECL3.
 | ||
| Fig. 7 The RMSF value of apo-GLP-1R and holo-GLP-1R (y-axis) in MD simulation for individual along residues (x-axis). | ||
We analyzed the structural rearrangements of GLP-1R upon activation caused by GLP-1 by superposition of the middle structures of the biggest cluster for 200–220 ns trajectories of apo-GLP-1R and GLP-1/GLP-1R complex (Fig. 8). The ECD and the loop linking the ECD and the TMD of the apo-GLP-1R structure adopt a conformation that can partially block the binding of GLP-1 to GLP-1R. The ECL3, the ECD and the linking loop of apo-GLP-1R were gathering much closer than those of GLP-1/GLP-1R complex, allowing TM5 and TM6 to move outward simultaneously. However, ECL1 moved farther from the center of TMD when the ECD of GLP-1R fell down toward the transmembrane domain in the apo system. And this is consistent with MD simulations of apo-GCGR, in which ECL1 of apo-GCGR also deviates farther from the center of TMD. Furthermore, it can be observed obviously that ECL3 and the extracellular part of TM6 and TM7 in the complex moved outward to accommodate GLP-1 with respect to apo-GLP-1R. Comparing GLP-1/GLP-1R complex with apo-GLP-1R, the binding pocket formed by TM1, TM2, TM3, TM5, TM6, TM7, ECL2 and ECL3 of GLP-1R specially expanded for accommodating GLP-1. The distance between the COMs (center of mass) of the ECD and TMD (Fig. 9) in the holo-GCGR is distinctly larger than that of apo-GLP-1R, suggesting apo-GCGR and holo-GLP-1R maintain two different states.
 | ||
| Fig. 8 Superposition of the middle structures of the biggest cluster for 200–220 ns trajectories of apo-GLP-1R (yellow) and GLP-1/GLP-1R complex system (GLP-1R is colored purple and GLP-1 is colored grey), representing closed and open state, respectively. | ||
 | ||
| Fig. 9 The evolution of the distances between the COMs (center of mass) of the ECD and TMD in holo-GCGR (red line) and apo-GLP-1R (black line). | ||
The apo-GLP-1R adopts a conformation with the ECD covering the helix bundle of the transmembrane domain, representing a kind of ‘closed’ state, just like a box shutting lid. On the contrary, the GLP-1R in the complex adopts a conformation to accommodate GLP-1, representing a kind of ‘open’ state, just like a box opening lid. Our MD simulation results provide solid evidences for the existence of the closed state of GLP-1R. In the apo system, we found that snapshots of 15–35 ns mainly adopted an open conformation and snapshots of 200–220 ns mainly adopted a closed conformation. The total potential of GLP-1R in the closed state (−13544.0 kJ mol−1) was much lower than that of GLP-1R in the open state (−10374.6 kJ mol−1), indicating that GLP-1R prefers to stabilize at the closed state when no ligand is present. This result is in line with the putative closed state of GCGR proposed by Linlin Yang et al. According to their study, apo-GCGR can adopt both open and closed states, while the closed state is energetically more accessible due to extensive contacts between the ECD and TMD. We suggested that apo-GLP-1R can adopt conformations corresponding to the open or the closed state and two kinds of states maintain a balance. Though the conformation corresponding to the closed state is more energetically favorable, GLP-1 prefers to bind to the GLP-1R in the open state, inducing the equilibrium of the system changing from a closed conformation dominated state to an open state dominated state. The activation mechanism of GLP-1 was closely associated with the intrinsic conformational change of the GLP-1R structure. Considering the high resemblance between GCGR and GLP-1R, they might share some common characteristics in the signal transduction mechanism, as well as the other class B GPCRs. The proposed binding mechanism of GLP-1 to GLP-1R can also be applied to other GPCRs in class B.
Our GLP-1/GLP-1R complex model agrees well with ‘two-domain’ ligand binding model, the C-terminal region of the GLP-1 interacts with the ECD of GLP-1R, and the N-terminal region of GLP-1 locating in the pocket formed by the residues in the transmembrane helical bundles and ECLs. In fact, GLP-1 has conserved features with its family peptides, for example, H1, G4, T7 and E9 of GLP-1 exist likewise in glucagon, GCRP and GLP-28. Therefore, it was speculated that GLP1R, GCGR, and GCRPR may share similar ligand-binding pockets.
3.4 The dynamic nature of GLP-1
It can be found that NMR structures of GLP-1 (PDB ID 1D0R) exist in two major forms, namely, a straight helix or a bent helix similar to L-shape. It is not clear which form is adopted or more stable in GLP-1R/GLP1 complex. In previous work,36 there was discussion about this issue and the L-shaped was preferred. However, it was observed that GLP-1 with a L-shaped helix as the starting structure gradually turned into conformation with a stable and nearly straight helix during MD simulation in our work. The structure of GLP-1 adopted by the NMR structures (PDB ID 1D0R) consists of a little bent helix from V10 to V27 and two random coils of C terminal and N terminal, similar to L shape. And Q17 is positioned adjacent to helix turn. Nevertheless, the helix from V10 to V27 kept straight in the last equilibrium stage. To better describe the conformation change of GLP-1, the angle of the helical buckling was defined as the angle composed of three Cβ atoms of V10, Q17 and V27 with the Cβ atom of Q17 as the turning point. The fluctuation of such angle is depicted in Fig. 10, where the angle reaches a stable state at about 60 ns. Helical buckling of GLP-1 from 20 to 40 ns and from 200 to 220 ns is stabilized at about 120° and 160°, respectively. In Linlin Yang's work,41 they proposed that a large conformational change of GCGR from the closed state to open state would need a tensile force exerted by an immobilized ligand.42 A straight helix is a less flexible but more fixed conformation, and herein, we suggest that a more fixed conformation of GLP-1 might be favorable to realize conformational change of GLP-1R from the closed state to open state. Certainly, more studies including validation experiment need to be done later. | ||
| Fig. 10 The angle of helical buckling along the time frame for GLP-1. The angle of the helical buckling was defined as the angle composed of three Cβ atoms of V10, Q17 and V27 with the Cβ atom of Q17 as the turning point. | ||
4. Conclusions
In the present work, based on the crystal structures of GCGR and ECD structure of GLP-1R coupled with GLP-1, the complete model of GLP-1/GLP-1R complex was constructed by means of homology modeling and precise protein–protein docking. Subsequently, long-time MD simulations were performed for both apo-GLP-1R and GLP-1/GLP-1R complex systems. The binding mode between GLP-1 and GLP-1R was depicted in detail. It is encouraging that some key residues in our GLP-1/GLP-1R complex structure responsible for ligand–receptor interaction correspond to mutation experiments and previous work. In addition, a big conformational difference between the apo-GLP-1R and GLP-1/GLP-1R was observed. That is the apo-GLP-1R is more likely to adopt a kind of closed state when the ligand is absent, whereas GLP-1-bound GLP-1R maintains a kind of open state. According to the MD results, it was inferred that GLP-1 is likely to bind to GLP-1R by a more fixed conformation exerting a tensile force. Our model and findings may not only help understand the binding mode of GLP-1 to GLP-1R and the relative dynamic nature between ECD and TMD of GLP-1R, but also supply useful information for anti-diabetic drug design.Acknowledgements
We gratefully acknowledged the financial supports from the 863 Project (No. 2012AA020308), National Natural Science Foundation of China (No. 81273438 and 81373329), and the Innovation Program of Shanghai Municipal Education Commission (No. 13ZZ044).References
- K. Hollenstein, C. D. Graaf, A. Bortolato, M. W. Wang, F. H. Marshall and R. C. Stevens, Trends Pharmacol. Sci., 2014, 35, 12–22 CrossRef CAS PubMed.
- A. Bortolato, A. S. Doré, K. Hollenstein, B. G. Tehan, J. S. Mason and F. H. Marshall, Br. J. Pharmacol., 2014, 171, 3132–3145 CrossRef CAS PubMed.
- S. M. Lee, J. M. Booe and A. A. Pioszak, Eur. J. Pharmacol., 2015, 763, 196–205 CrossRef CAS PubMed.
- T. J. Kieffer and J. F. Habener, Endocr. Rev., 1999, 20, 876–913 CrossRef CAS PubMed.
- M. E. Doyle and J. M. Egan, Pharmacol. Ther., 2007, 113, 546–593 CrossRef CAS PubMed.
- L. L. Baggio and D. J. Drucker, Gastroenterology, 2007, 132, 2131–2157 CrossRef CAS PubMed.
- M. Sulistio, C. Carothers, M. Mangat, M. Lujan, R. Oliveros and R. Chilton, Curr. Atheroscler. Rep., 2009, 11, 93–99 CrossRef CAS PubMed.
- J. I. Hwang, S. Yun, M. J. Moon, C. R. Park and J. Y. Seong, J. Mol. Endocrinol., 2014, 52, T15–T27 CrossRef CAS PubMed.
- D. I. Buckley, J. F. Habener, J. B. Mallory and S. Mojsov, US Pat., 5545618A, 1996.
- M. A. Nauck, J. J. Holst, B. Willms and W. Schmiegel, Exp. Clin. Endocrinol. Diabetes, 1997, 105, 187–195 CrossRef CAS PubMed.
- L. B. Knudsen, P. O. Huusfeldt and P. F. Nielsen, US Pat., 8097698B2, 2012.
- D. K. Arulmozhi and B. Portha, Eur. J. Pharm. Sci., 2006, 28, 96–108 CrossRef CAS PubMed.
- L. B. Knudson, P. O. Huusfeldt and P. F. Nielsen, US Pat., 7235627B2, 2007.
- B. Ahrén, Eur. Diabetes Nurs., 2013, 10, 31–36 CrossRef.
- D. M. Kendall, M. C. Riddle, J. Rosenstock, D. Zhuang, D. D. Kim, M. S. Fineman and A. D. Baron, Diabetes Care, 2005, 28, 1083–1091 CrossRef CAS PubMed.
- D. C. Klonoff, J. B. Buse, L. L. Nielsen, X. Guan, C. L. Bowlus, J. H. Holcombe, M. E. Wintle and D. G. Maggs, Curr. Med. Res. Opin., 2008, 24, 275–286 CrossRef CAS PubMed.
- D. Sennik, F. Ahmed and D. Russell-Jones, Expert Rev. Endocrinol. Metab., 2014, 7, 15–26 CrossRef.
- L. B. Knudsen, P. F. Nielsen, P. O. Huusfeldt, N. L. Johansen, K. Madsen, F. Z. Pedersen, H. Thøgersen, M. Wilken and H. Agersø, J. Med. Chem., 2000, 43, 1664–1669 CrossRef CAS PubMed.
- J. B. Buse, J. Rosenstock, G. Sesti, W. E. Schmidt, E. Montanya, J. H. Brett, M. Zychma and L. Blonde, Lancet, 2009, 374, 39–47 CrossRef CAS.
- M. Lind, J. Jendle, O. Torffvit and I. Lager, Diabetes Prim. Care, 2012, 6, 41–46 CrossRef PubMed.
- F. Y. Siu, M. He, C. D. Graaf, G. W. Han, D. Yang, Z. Zhang, C. Zhou, Q. Xu, D. Wacker, J. S. Joseph, W. Liu, J. Lau, V. Cherezov, V. Katritch, M. W. Wang and R. C. Stevens, Nature, 2013, 499, 444–449 CrossRef CAS PubMed.
- K. Hollenstein, J. Kean, A. Bortolato, R. K. Y. Cheng, A. S. Doré, A. Jazayeri, R. M. Cooke, M. Weir and F. H. Marshall, Nature, 2013, 499, 438–443 CrossRef CAS PubMed.
- P. M. Sexton and D. Wootten, Nature, 2013, 499, 417–418 CrossRef CAS PubMed.
- C. R. Underwood, P. Garibay, L. B. Knudsen, S. Hastrup, G. H. Peters, R. Rudolph and S. Reedtz-Runge, J. Biol. Chem., 2010, 285, 723–730 CrossRef CAS PubMed.
- M. J. Moon, Y. N. Lee, S. Park, A. Reyes-Alcaraz, J. I. Hwang, R. P. Millar, H. Choe and J. Y. Seong, J. Biol. Chem., 2015, 290, 5696–5706 CrossRef CAS PubMed.
- K. Coopman, R. Wallis, G. Robb, A. J. H. Brown, G. F. Wilkinson, D. Timms and G. B. Willars, Mol. Endocrinol., 2011, 25, 1804–1818 CrossRef CAS PubMed.
- X. Chang, D. Keller, S. Bjørn and J. J. Led, Magn. Reson. Chem., 2001, 39, 477–483 CrossRef CAS.
- Q. Xiao, W. Jeng and M. B. Wheeler, J. Mol. Endocrinol., 2000, 25, 321–335 CrossRef CAS PubMed.
- R. López De Maturana and D. Donnelly, FEBS Lett., 2002, 530, 244–248 CrossRef.
- S. Al-Sabah and D. Donnelly, FEBS Lett., 2003, 553, 342–346 CrossRef CAS PubMed.
- X. Sun, H. Ågren and Y. Tu, J. Phys. Chem. B, 2014, 118, 14737–14744 CAS.
- X. Sun, J. Cheng, X. Wang, Y. Tang, H. Ågren and Y. Tu, Sci. Rep., 2015, 5, 8066 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. V. D. Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- P. L. Brubaker and D. J. Drucker, Recept. Channels, 2002, 8, 179–188 CrossRef CAS PubMed.
- F. Lin and R. Wang, J. Mol. Model., 2009, 15, 53–65 CrossRef CAS PubMed.
- S. Mojsov, G. Heinrich, I. B. Wilson, M. Ravazzola, L. Orci and J. F. Habener, J. Biol. Chem., 1986, 261, 11880–11889 CAS.
- C. Sarrauste De Menthière, A. Chavanieu, G. Grassy, S. Dalle, G. Salazar, A. Kervran, B. Pfeiffer, P. Renard, P. Delagrange and D. Manechez, Eur. J. Med. Chem., 2004, 39, 473–480 CrossRef PubMed.
- B. Manandhar and J. M. Ahn, J. Med. Chem., 2015, 58, 1020–1037 CrossRef CAS PubMed.
- D. Wootten, J. Simms, L. J. Miller, A. Christopoulosa and P. M. Sexton, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5211–5216 CrossRef CAS PubMed.
- L. Yang, D. Yang, C. D. Graaf, A. Moeller, G. M. West, V. Dharmarajan, C. Wang, F. Y. Siu, G. Song, S. Reedtz-Runge, B. D. Pascal, B. Wu, C. S. Potter, H. Zhou, P. R. Griffin, B. Carragher, H. Yang, M. W. Wang, R. C. Stevens and H. Jiang, Nat. Commun., 2015, 6, 7859 CrossRef CAS PubMed.
- L. J. Miller, Q. Chen, P. C. H. Lam, D. I. Pinon, P. M. Sexton, R. Abagyan and M. Dong, J. Biol. Chem., 2011, 286, 1375–1381 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |