
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aggregation of layered double hydroxide nanoparticles in the presence of heparin: towards highly stable delivery systems†
Marko
Pavlovic
a,
Li
Li
b,
Francois
Dits
a,
Zi
Gu
b,
Monika
Adok-Sipiczki
a and
Istvan
Szilagyi
*a
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, Geneva, CH-1205, Switzerland. E-mail: istvan.szilagyi@unige.ch; Tel: +41 22 3796031
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD-4072, Australia
First published on 4th February 2016
Abstract
The effect of heparin adsorption on the colloidal stability of layered double hydroxide particles as potential drug delivery agents was studied in aqueous suspensions. The lamellar structures were prepared by the co-precipitation method and composed of magnesium(II) and aluminium(III) mixed hydroxide as the layers and carbonate anions between the layers. Stable and positively charged particles were observed at low heparin concentrations and low ionic strengths where the surface charge was only partially neutralized by the oppositely charged natural polyelectrolyte adsorbed on the surface. Increasing the heparin dose resulted in charge neutralization and subsequent charge reversal at appropriate doses. The particles aggregated rapidly in the absence of sufficient surface charge, however, remarkably stable dispersions were obtained when the particles were completely covered by heparin. The latter coating process gave rise to two-times higher surface charge density in magnitude and about 20-times higher critical coagulation concentration than for the bare particles. The significant stabilization effect due to the heparin-coating resulted from repulsive interparticle forces of electrostatic and steric origin. On the basis of these findings, efficient delivery systems can be designed where the colloid stability of the carrier particles is enhanced by coating with a biocompatible polyelectrolyte.
Introduction
Inorganic nanoparticles have been widely used as efficient gene and drug delivery agents.1–5 Among them, layered double hydroxides (LDHs) are popular nanocarriers due to their ionic exchange property which allows intercalation of negatively charged bioactive substances between the layers as well as adsorption on the surface.3,6–11 LDHs can be readily synthesized12,13 and their size can be tuned to achieve the desired cellular uptake and drug release in the delivery process.14–17 Similarly to other nanoparticles used in biomedical processes,18 the colloidal stability of LDH-based carriers is a critical issue, since aggregation of the particles can prevent the successful delivery of the target molecules. Despite its importance, stabilization of LDH particles in aqueous dispersions has not been yet investigated in detail and the first systems describing highly stable protein or polymer modified platelets have been reported only recently.19–21Polyelectrolytes have been proved as effective stabilizing agents for nanoparticles to be used in biochemical processes.22–24 In particular, low molecular weight heparin, a biocompatible sulfated polysaccharide which can be considered as natural polyelectrolyte, is a powerful injectable anticoagulant for numerous biomedical applications, because of its highest negative line charge density among the known biological molecules.25 The effect of heparin adsorption on stability of nanoparticles has been investigated in certain systems. For instance, aggregation of gold nanoparticles was found to be sensitive to the heparin concentration.26 This discovery led to the development of optical sensor to determine the concentration of the macromolecules based on the different visible spectra of the gold aggregates of different size.27,28 Aggregation of drug nanocapsules was also prevented by using heparin during synthesis,29 and nanogels composed of heparin and positively charged polyelectrolytes were used successfully in gene delivery for cancer treatment.30 Apart from the applications as stabilizing agent or building block for carrier nanomaterials, heparin was also delivered as drug in biomedical treatments.31,32
Loading LDH with heparin has been studied mainly from the point of view of intercalation between the LDH layers. This type of composite materials can be used as drugs for clinical applications.33 Such immobilization and the resulted structure of the nanocomposite have been investigated by both experimental34 and theoretical35 methods. Accordingly, successful intercalation was reported among the layers if heparin was present together with the chloride salts of magnesium(II) and aluminium(III) ions in the co-precipitation process where the pH was increased to form heparin-LDH lamellar material.34 A computational modelling study revealed that attractive electrostatic forces were responsible for the adsorption and strong immobilization on the lamellae and the heparin helices were oriented in parallel with the LDH layers due to structural deformation of the polyelectrolytes upon intercalation.35 Self-diffusion of water and heparin and kinetics of drug release were also addressed in these papers. In addition, ultrathin biomimetic film composed of delaminated LDH nanosheets and heparin layers was prepared by the layer-by-layer method utilizing the high affinity of the heparin to the oppositely charged platelets.36 The obtained film showed enhanced strength and good blood biocompatibility which can be beneficial in certain medical applications. Apart from these papers, studies on colloid stability of LDH particles in the presence of heparin have not been reported so far.
Therefore, in this research we carried out the investigation on the effect of heparin adsorption on aggregation of LDH particles of different sizes in detail. First of all, charging and stability of LDH particles, composed of magnesium(II) and aluminium(III) hydroxide and carbonate anions in the gallery, were studied in aqueous dispersions without and with heparin. The lamellar structure of the particles was determined by X-ray diffraction (XRD) and the colloidal properties were explored in electrophoretic and time-resolved dynamic light scattering (DLS) experiments. The orientation of the particles in stable and aggregated samples was finally examined by transmission electron microscopy (TEM). The results of the present work revealed that the speed of particle aggregation and related dispersion stability of the LDH-heparin colloids can be tuned by changing the experimental conditions such as polyelectrolyte dose, particle concentration and ionic strength. In this way, highly stable samples can be designed and applied as biocompatible carrier systems in further biomedical applications, especially in drug and gene delivery processes.
Experimental section
Materials
Low molecular weight sodium heparin was purchased from Acros Organics and used for sample preparation without further purification. The manufacturer reported an average molecular weight of about 13.5 kg mol−1. Other chemicals such as MgCl2, AlCl3, Na2CO3, NaOH, HCl and NaCl (all from Sigma-Aldrich) used in the experiments were analytical grade or higher quality. Ultrapure water (Millipore) was used for solution preparations which were adjusted to pH (7.0 ± 0.5) for all stock solutions and dispersions before mixing them together. The experiments were carried out at a temperature of (25 ± 0.2) °C.LDH particles were prepared by the co-precipitation method followed by hydrothermal treatment.37–40 Briefly, a mixture of 0.6 M MgCl2 and 0.2 M AlCl3 solution (10 mL) was quickly added to 40 mL of mixed solution containing 0.40 M NaOH and 0.04 M Na2CO3 under vigorous stirring. After mixing for 10 min, the LDH slurry was collected and washed twice. Then the particles were manually re-suspended in 40 mL deionized water. The inhomogeneous suspension was transferred to an autoclave (stainless steel with Teflon lining) and heated in an oven at 100 °C for 16 h (sample LDH1) or at 150 °C for 16 h (LDH2). After this hydrothermal treatment, a transparent and homogenous LDH suspension was obtained with the mass concentrations of 9.3 and 8.0 g L−1 for LDH1 and LDH2, respectively. To completely cover the nanoparticles with heparin, 100 mg macromolecule was adsorbed on 1 g of LDH under the desired experimental conditions, as detailed later.
Electrophoresis
The electrokinetic potential was converted from the electrophoretic mobility which was measured with a ZetaNano ZS (Malvern) device using an electric field of 4 kV m−1. The experiments were performed in plastic capillary cells (Malvern) cleaned with 2% Hellmanex (Hellma) solution and rinsed with ultrapure water. In each measurement, 5 mL dispersion was prepared as follows. A calculated amount of heparin stock solution was mixed with NaCl solution and water to obtain the desired polyelectrolyte dose and ionic strength. The sample preparation was finalized by adding 0.5 mL of the particle stock dispersion of about 93 and 80 mg L−1 (diluted from the original suspensions) for samples LDH1 and LDH2, respectively. The suspensions were equilibrated overnight at room temperature and their electrophoretic mobility was measured five times and the averaged. The method of conversion to electrokinetic potential is detailed in the ESI.†Light scattering
The aggregation of the LDH particles was followed in time-resolved DLS experiments which were carried out with a compact goniometer system (ALV/CGS-3) at 90° scattering angle. The instrument was equipped with a He/Ne laser operating at 633 nm as a light source and an avalanche photodiode as a detector. To determine the hydrodynamic radius (Rh) of the LDH particles, the correlation function was accumulated for 20 seconds and a second cumulant fit was used.41 The Rh measured for the platelet-like LDH particles is equal to the radius of an equivalent sphere. As shown in Fig. 1 for some examples, the Rh values were recorded in time-resolved measurements for 15–20 minutes depending on the speed of the aggregation. The increase in Rh was always linear in these experiments, indicating early stages of the aggregation. The slopes in the Rhversus time plots were used to calculate the stability ratio (W) via the following equation:42–44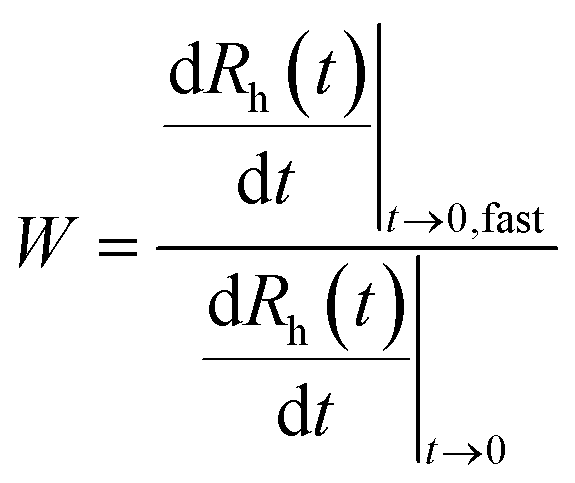 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and washed with water followed by drying in a dust-free oven. Further information on the DLS measurements can be found in the ESI.†
:1 and washed with water followed by drying in a dust-free oven. Further information on the DLS measurements can be found in the ESI.†
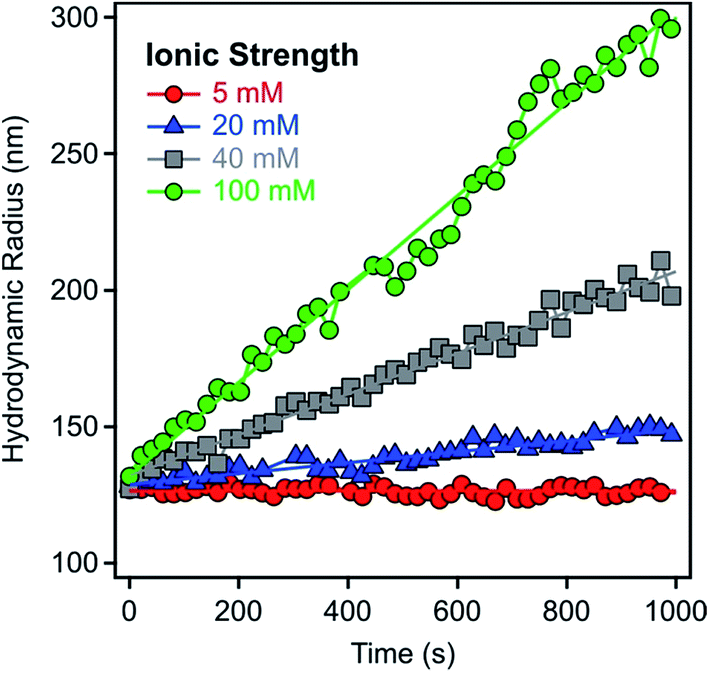 | ||
| Fig. 1 Hydrodynamic radii of LDH1 particles versus time measured in time-resolved DLS experiments at 9.3 mg L−1 particle concentration and different ionic strengths adjusted with NaCl. | ||
X-ray diffraction
Powder XRD patterns of solid LDH samples were registered in the 2-theta range of 3–80° on a Stadi-P (Stoe) instrument. This device uses CuKα (λ = 0.15406 nm) radiation in Bragg–Brentano geometry. Positions of the peaks and full widths at half maximum (FWHM) were determined by applying Gaussian fits. More details about the interpretation of the XRD spectra are provided in the ESI.†Transmission electron microscopy
TEM imaging was carried out with a Tecnai G2 electron microscope (FEI) operating at 120 kV. During sample preparation, 3 μL of the particle dispersion was placed on carbon coated 400-mesh copper grids, left for 30 seconds and drained off the excess liquid with filter paper and dried. The same experimental conditions (e.g., particle concentration, pH and heparin dose) were applied for the original TEM samples as those used in electrophoretic and aggregation studies.Results and discussion
Particle characteristics
We observed seven different peaks in the XRD pattern of both LDH1 and LDH2 (Fig. 2). Comparing them to the diffraction pattern of other carbonate LDHs,45–47 we can unambiguously confirm the formation of the lamellar structure typical for this kind of LDHs. However, the minor peaks observed at 19° indicates the presence of a small amount of Mg(OH)2 side phase. We could assign Miller indexes to each peak and use some of them to calculate lattice parameters which are necessary to describe a unit cell (Table 1). From the position of the first peak 003, a d003 parameter of 0.77 nm was calculated for both LDH particles. This value represents the thickness of one hydroxide layer and one interlayer spacing together. From the d003 value, the unit cell (c = 2.31 nm) was calculated for LDH1 and LDH2. Another important lattice parameter is the shortest distance between two cations in one hydroxide layer expressed by parameter a, which can be calculated as twice the d-spacing of the 110 reflection. The same a value (0.31 nm) was obtained for both particles. In addition, the average thickness of the particles (υ) along the c axis was found to be 18.2 and 17.3 nm for LDH1 and LDH2, respectively. This implies that the particles were constituted by about 20–25 brucite-like hydroxide layers. DLS measurements yielded hydrodynamic radii of 136.1 and 316.2 nm in stable dispersions for LDH1 and LDH2, respectively. The particles were slightly polydisperse, as indicated by their polydispersity index (PDI) values determined in the DLS experiments (Table 1).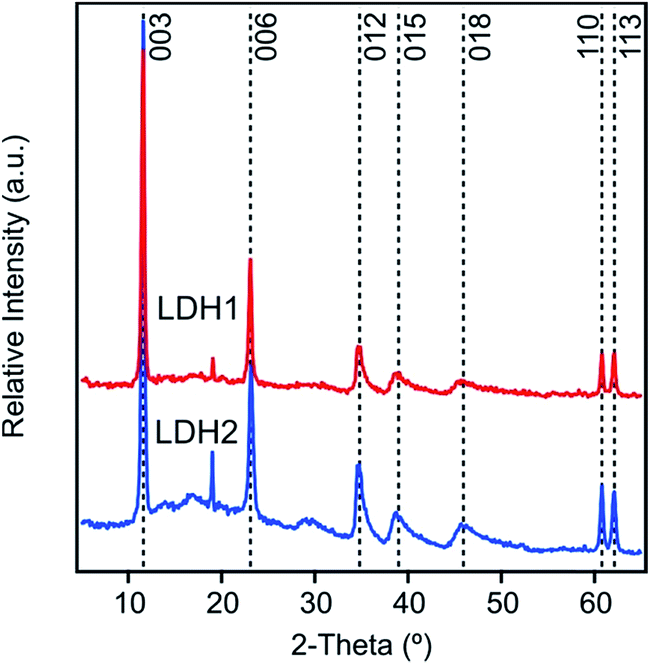 | ||
| Fig. 2 Powder XRD pattern of the LDH particles, composed of magnesium(II) and aluminium(III) cations and carbonate interlayer anions, synthesized by co-precipitation. The peak at 19° could be due to the 001 peak of Mg(OH)2 that was formed from soluble magnesium(II) and hydroxide ions after drying. | ||
| Peak positiona (°) | Calculated parameters (nm) | Sizeb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 003 | 110 | d 003 c | d 110 c | a d | c e | ν f | R h (nm) | PDI | |
| a Obtained from the XRD spectra. b Hydrodynamic radius and polydispersity index were measured by DLS. c Calculated with eqn (S8) (see ESI). d Shortest distance between two cations in the layer and a = 2d110. e The width of a unit cell and c = 3d003. f Average particle thickness, calculated with eqn (S9) (see ESI). | |||||||||
| LDH1 | 11.6 | 60.8 | 0.77 | 0.15 | 0.31 | 2.31 | 18.2 | 136.1 | 0.274 |
| LDH2 | 11.6 | 60.8 | 0.77 | 0.15 | 0.31 | 2.31 | 17.3 | 316.2 | 0.265 |
Charging of LDH in the presence of heparin
Surface charge of LDH1 was investigated first with electrophoresis. The particles possessed positive charges (electrokinetic potential was about 40 mV in aqueous dispersion without added polyelectrolyte at low ionic strength) due to their structural properties while the heparin is negatively charged at the pH used (7.0 ± 0.5). In the individual experiments, particle concentration, ionic strength and pH were kept constant while the heparin concentration was varied. Such measurements were repeated at different ionic strength (1, 10 and 100 mM adjusted by NaCl) to explore the adsorption mechanism.In general, the electrokinetic potentials were positive at low polyelectrolyte concentrations (0–1 mg g−1, and the mg g−1 unit refers to mg heparin per one gram of particle) indicating the positive charge of the bare platelets (Fig. 3a). Increasing the heparin dose, the potential values decreased due to the heparin adsorption on the oppositely charged surface. This process gave rise to charge neutralization at the isoelectric point (IEP) when the heparin concentration reached the range of 12–26 mg g−1 depending on the ionic strength applied. The IEP corresponds to the dose where the overall charge of the particle is zero. Further adding heparin to the samples, charge reversal48–50 occurred, indicating the continuous adsorption of heparin on the LDH1 platelet surface.51 Similar charge reversal has been already observed in several dispersions containing colloidal particles and polyelectrolytes21,22,52–55 or multivalent ions56–58 and also in other systems of similar charge balance.29,59 Monovalent anions can also cause such charge reversal of LDHs, but in a much smaller extent indicating the significantly higher affinity of the negatively charged polyelectrolytes (e.g., heparin) to the oppositely charged particle surface.
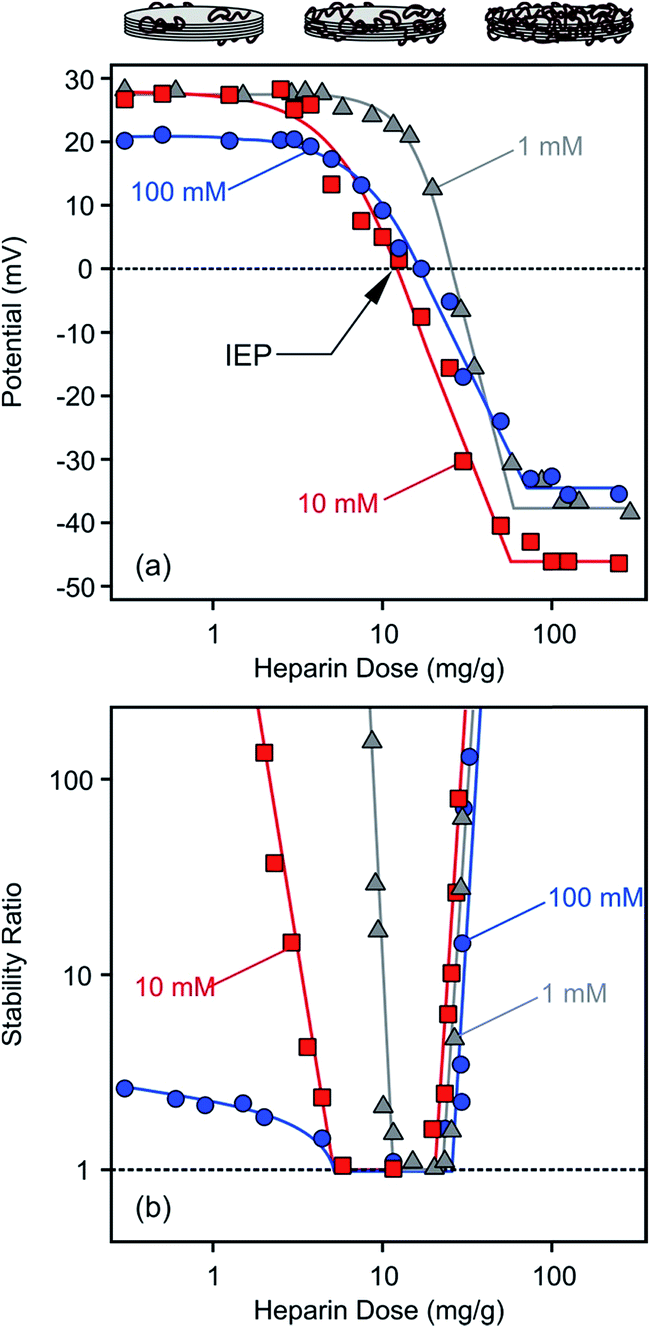 | ||
| Fig. 3 Electrokinetic potential (a) and stability ratio (b) values for LDH1 in the presence of heparin at different ionic strengths. Stability ratios close to unity indicate rapid particle aggregation while higher values point to more stable dispersions. The mg g−1 unit refer to mg heparin per gram of LDH. The lines are just to guide the eyes. | ||
The potential versus heparin dose curves saturated at high concentrations indicating full coverage of the LDH particle surface with the polyelectrolyte under these experimental conditions. Additional heparin molecules remained dissolved in solution after this concentration. Such saturation took place around 100 mg g−1 polyelectrolyte dose regardless of the ionic strength. However, the magnitude of the electrokinetic potential was sensitive to the ionic strength. At low heparin concentrations (0–3 mg g−1), where the positive charge of the particles was only partially compensated by the adsorbed molecules of opposite charge, the potential values decreased at higher salt level (100 mM) due to the screening effect of the counterions on the surface charge. Once the heparin dose was high enough (100–300 mg g−1), the ionic strength dependence of the potentials reflected the surface charge characteristics of the coated LDH1 at different salt levels as discussed later. Multivalent organic anions containing carboxylate groups showed similar adsorption properties on carbonate and chloride LDHs.51 Accordingly, charge neutralization and subsequent charge reversal were reported on the basis of zeta potential measurements, but such processes took place in a much higher concentration range in those measurements due to the higher particle concentrations applied.
Aggregation in the presence of heparin
Time-resolved DLS experiments were performed to follow the aggregation processes under the same experimental conditions as in the charge investigation above. At a low ionic strength (1 mM), stability ratios close to unity indicated unstable dispersions near the dose corresponding to the IEP (Fig. 3b). The aggregation slowed down, i.e., the stability ratios increased, at low and high heparin doses. This behaviour can be well-explained by the classical theory developed by Derjaguin, Landau, Verwey and Overbeek (DLVO).60 Accordingly, the repulsive electrical double layer forces between the particles vanish in the absence of surface charge and the attractive van der Waals forces will destabilize the particle colloids at the IEP. Significantly below or above this dose, the particles have sufficient charge (Fig. 3a) and hence, electrical double layers form around the LDH, giving rise to repulsive forces and more stable dispersions. Such U-shaped curves are typical for polyelectrolyte-induced particle aggregation and found earlier in case of latex,61 titanate21,62,63 or silica64,65 colloids in the presence of oppositely charged macromolecules.The effect of ionic strength on particle aggregation was more pronounced at low heparin doses. The relatively narrow stability ratio versus dose curve at 1 mM salt level became larger at 10 mM. The dispersions possessed only limited stability at 100 mM NaCl, as indicated by stability ratios less than 3 (meaning that each third collision of the particles results in dimer formation) at very low heparin concentrations. The stability ratio values overlap within the experimental error at high doses and the transition between slow and fast aggregation was very abrupt (i.e., the slopes were very steep) in this regime indicating that the charge reversal-induced restabilization of the dispersions is not very sensitive to the ionic strength. The dependence of the aggregation rates on the salt level is in line with the DLVO theory and can be explained with screening the surface charge with the counterions, which leads to smaller Debye lengths and weaker repulsive double layer forces at high salt levels. However, the heparin coated LDH1 was stable even at high ionic strengths, indicating an extremely high surface charge density or the presence of additional repulsive forces of non-DLVO origin.66–68 This issue will be further explored shortly.
The stability of the dispersions and the structure of aggregates were studied by recording TEM images with the same samples used for the electrophoretic and time-resolved DLS experiments. Such images were taken at the low heparin dose (Fig. 4a), near the IEP (Fig. 4b) and at the high polyelectrolyte concentration (Fig. 4c). In the last case, the LDH1 particles should be completely covered by heparin. Note that the samples were dried during the TEM images, which could also induce some aggregation. Nevertheless, well-separated particles were observed at the low heparin dose indicating stable samples. Aggregation was observed close to the IEP, in good agreement with the stability study. Similar to other LDH systems,69,70 the platelets prefer face-to-face orientation in the aggregates. Other types of orientation such as face-to-edge71 or edge-to-edge72,73 were not observed in the TEM images. Stable samples and separated particles were imaged at the high heparin dose where the aggregation was prevented by the adsorbed polyelectrolyte layer.
 | ||
| Fig. 4 TEM images of LDH1 particles at different heparin doses: 1 mg g−1 which refers to low dose and stable samples (a), 15 mg g−1 near the IEP with aggregated samples (b) and 30 mg g−1 where the particles reversed their charge and form stable dispersion (c). | ||
Effect of particle size
The changing trend in potentials (Fig. 5a) and stability ratios (Fig. 5b) was found to be similar for LDH2 particles of larger size by increasing the heparin concentration, indicating similar adsorption mechanism and interparticle forces as for the LDH1 of smaller size. However, differences can still be observed if one interprets the ionic strength dependence. At 100 mM NaCl concentration, the charge of the particles was close to zero at low heparin doses due to the enhanced screening effect of the counterions on the surface charge. Such low potentials led to unstable samples, indicated by stability ratios close to one in this regime. Similarly to the LDH1 system, the aggregation rates were sensitive to the electrolyte concentration mostly at low heparin doses, while stable dispersions were obtained at higher polyelectrolyte concentrations irrespectively from the ionic strength. TEM images, recorded with stable and unstable samples, confirm these findings (see Fig. S1 in ESI†). Accordingly, primary particles were found at low (1 mg g−1) and high (30 mg g−1) doses, while aggregates were imaged near the IEP (5 mg g−1). The LDH2 platelets also show face-to-face orientation in the aggregates.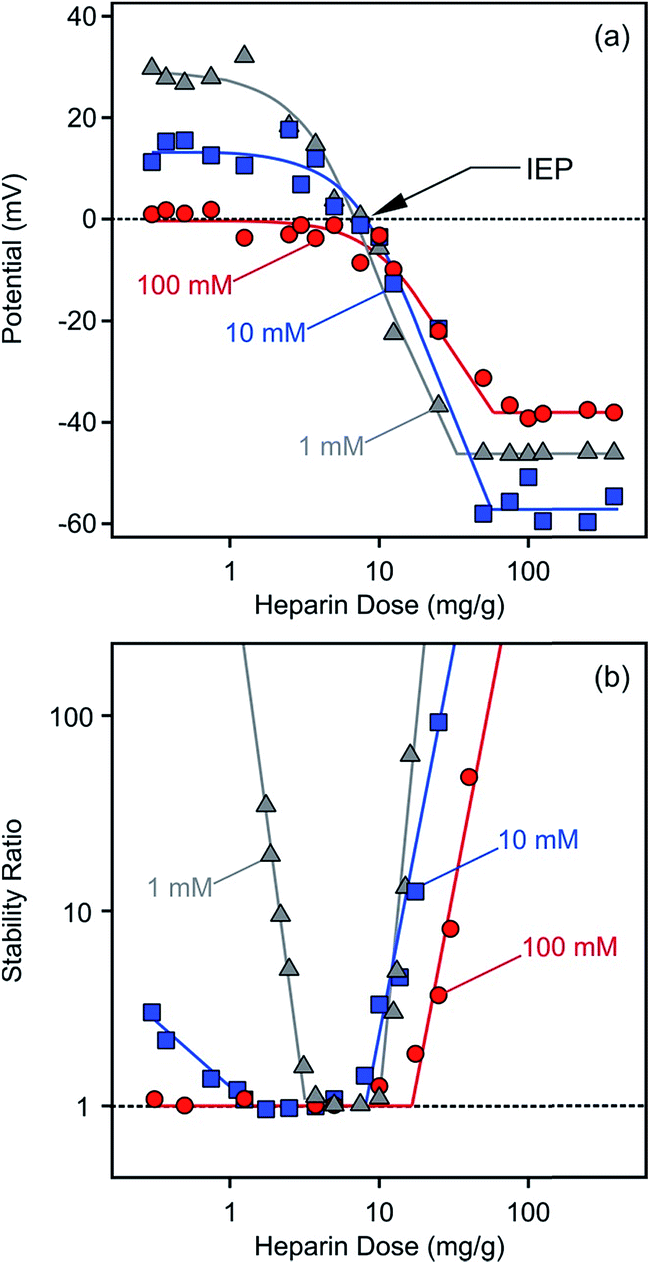 | ||
| Fig. 5 Electrokinetic potential (a) and stability ratio (b) values for LDH2 in the presence of heparin at different ionic strengths. The mg g−1 unit refer to mg heparin per gram of LDH. The lines are just to guide the eyes. | ||
Although intercalation of heparin between the layers of certain LDHs has been published earlier,34 we did not find any evidences for such process in our experiments. This is most likely due to the different conditions used in the reported study and our systems. The heparin is immobilized between the lamellae through the competition with chloride anions in the co-precipitation process in the report (where heparin has much higher affinity for LDH than chloride). However, the intercalation could occur only through exchange with carbonate (with high affinity for LDH) in our samples where it is also unlikely to introduce bulky macromolecules into the lamellar structure.
Stability of bare and coated particles
The effect of heparin coating on colloidal stability was explored by comparing charging and aggregation of bare and fully covered particles at different salt levels. To achieve such coating, a heparin dose of 100 mg g−1 was applied for both particles, since this concentration was slightly above the saturation limit observed in the electrophoretic mobility curves (Fig. 3a and 5a). The ionic strength dependence on the electrokinetic potentials and stability ratios were determined over a wide range of NaCl concentration (Fig. 6).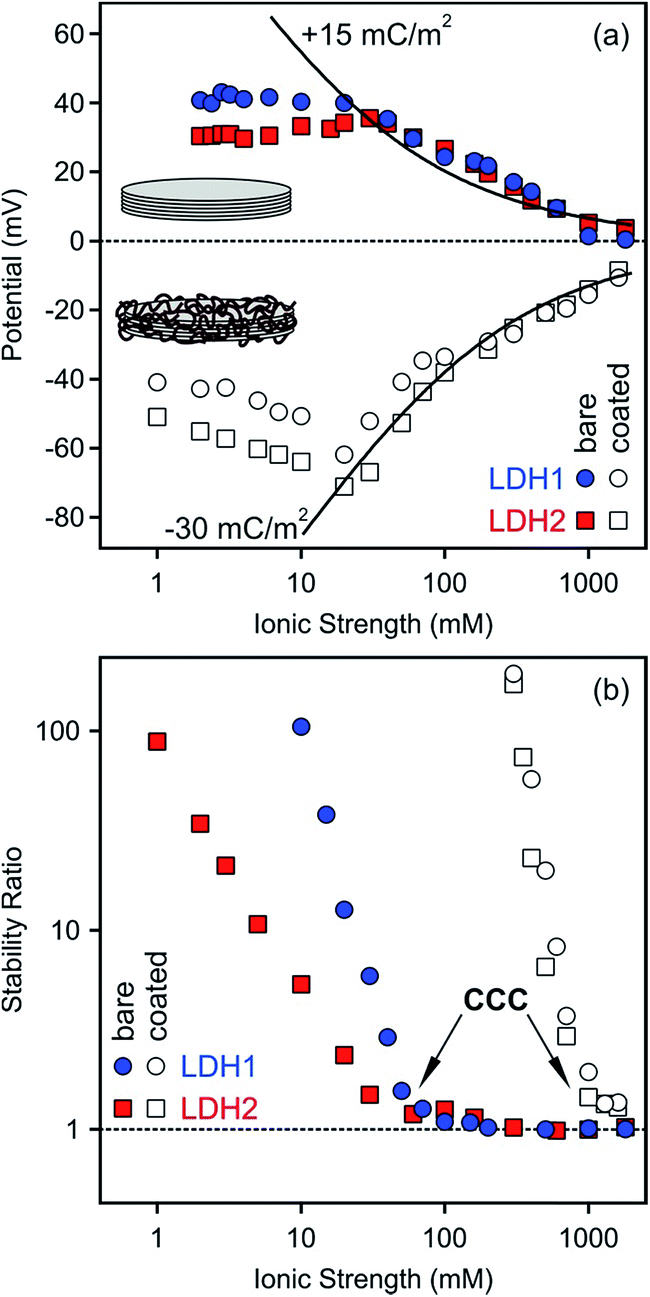 | ||
| Fig. 6 Electrokinetic potential (a) and stability ratio (b) values for the bare (filled symbols) and heparin coated (empty symbols, at a dose of 100 mg g−1) LDH1 and LDH2 particles at different ionic strengths. The solid lines in (a) indicates the potentials calculated using the Debye–Hückel model. The surface charge densities are also indicated. | ||
Slightly different potentials were obtained at low ionic strengths for the bare LDH1 and LDH2 particles, but they were the same within the experimental error at higher salt levels (Fig. 6a). The electrokinetic potentials decreased with the ionic strength due to the screening effect of the counterions on the particle surface and remained positive, although very close to zero at the higher electrolyte concentrations, in the entire concentration range investigated. This finding is in line with the results of our other study, in which we found that Cl− ions adsorb only weakly on LDHs, while more hydrophobic monovalent anions showed higher affinity and stronger adsorption on the surface.74
Heparin adsorption at the dose of 100 mg g−1 resulted in negatively charged particles as predicted by the results of the previous section. Similarly to the bare LDHs, the potentials of the coated ones deviated at low salt levels, while they showed good agreement at higher NaCl concentrations where the screening effect on the surface charges became important. Note that the sodium ions were the counterions in these systems. The effect of ionic strength on the potentials was similar again, namely, they decreased with increasing the salt concentration, but remained negative even at high electrolyte levels. Such change in the potential or related electrophoretic mobility is typical for bare or polyelectrolyte coated colloidal particles of significant charge.62,75 Theoretical potentials were calculated using the Debye–Hückel approximation (eqn (S2) in ESI†).60,62 This theory is valid at high ionic strengths and significant differences can be observed at lower salt levels. The best fit to the experimental values was found with +15 and −30 mC m−2 surface charge densities for the bare and heparin coated particles, respectively. These data indicate that the polyelectrolyte coverage resulted in two-time higher surface charge in magnitude compared to the LDH1 and LDH2 bare particles.
Stability ratios determined at different ionic strengths for the bare and coated particles showed similar trend in general (Fig. 6b). Accordingly, the dispersions were stable at low NaCl levels, while stability ratios close to one indicated rapidly aggregating dispersions at higher ionic strengths. The slow and fast aggregation regimes were separated by the well-defined critical coagulation concentration (CCC) value. Such behaviour is typical for charged colloidal particles and can be explained by the DLVO theory. The interparticle forces are the superposition of attractive van der Waals and repulsive double layer forces and the latter ones decrease with the ionic strength due to the progressively screened electrical double layer around the particles, leading to unstable particle dispersions at high salt levels.
Some differences can be observed in the stability ratio versus ionic strength curves for the bare particles. The CCC values felt in the same range, but slightly deviated (60 and 39 mM for LDH1 and LDH2, respectively). Such values are similar to those determined for LDH particles in the presence of simple monovalent anions (e.g., Cl− and NO3−), which are the counterions in these systems. However, complex and more hydrophobic anions (e.g., SCN−) tend to adsorb stronger on the particles and shift the CCC towards lower concentrations.74 In addition, the slopes in the slow aggregation regime are significantly different. For larger LDH2 particles, the stability ratios were smaller at the same NaCl concentrations, leading to smaller slopes at low ionic strengths compared to LDH1. Such deviation can be originated from the different sizes, since the attractive van der Waals forces are proportional to the Hamaker constant and the size of the particles.52,56,60 The Hamaker constant is related to the composition of the materials and hence, should be the same for both particles. However, the size dependence on the strength of the attractive van der Waals forces results in a stronger attraction for larger particles, leading to a lower CCC and a lower stability ratio before the CCC, in good agreement with the present results obtained with the bare systems.
For the heparin coated particles, the stability curves agreed within the experimental error and a common CCC around 1100 mM ionic strength was determined. Note that the sodium ions are the counterions in this system and they are responsible for the destabilization by electrostatic screening of the surface charge. The effect of monovalent cations on aggregation of colloidal particles is usually very small or negligible.76 This value is about 20-time larger than that for the bare LDHs, indicating an enormous stabilization effect of the heparin layer adsorbed on the platelets. Such polyelectrolyte-induced stabilization has been reported earlier for LDH20,21 and other colloidal particles.24,75,77,78 Since the size of the coated LDH1 particles is smaller than the LDH2 covered by heparin, different van der Waals forces can be expected and they should lead to different CCCs and slopes in the slow aggregation regime, similar to the bare particles discussed above. Nevertheless, the situation here is obviously different. The shape of the stability curves definitely indicates the presence of DLVO-type forces, but the extremely high CCC and the lack of differences in the stability ratios at lower ionic strength should be the sign of other types of repulsive interparticle forces. Polyelectrolyte layers adsorbed on oppositely charged surfaces are typically flat at low salt levels, but they are swollen at higher electrolyte concentrations.79 The swelling leads to the formation of polyelectrolyte tails and loops around the particles and steric repulsion between the hairy particles can become important.66,68,80 Our results can also be explained with the presence of repulsive steric forces between the adsorbed polyelectrolyte chains which are the most pronounced at high ionic strength due to the extended heparin layer around the platelets. As a result, these interparticle forces give rise to highly stable dispersions, which are potential candidates as carriers in delivery processes even at high salt levels where the bare particles would rapidly aggregate. In particular, the polyelectrolyte covered particles are stable at the physiological ionic strength (150 mM in blood), which is higher than the CCCs of the bare particles. Accordingly, intercalation of the lamellar particles with a desired drug molecule and coating with the appropriate dose of heparin will certainly result in delivery systems which can pass the critical part of living organisms without any unwanted aggregation of the carrier nanoplatelets.
Conclusions
Electrophoretic experiments revealed that heparin adsorption on oppositely charged LDH particles led to charge neutralization at the IEP and subsequent charge reversal at appropriate polyelectrolyte concentrations. The latter process led to two-time higher surface charge density in magnitude compared to the bare platelets. The electrokinetic potentials were sensitive to the ionic strength especially at low heparin doses due to the screening effect of the counterions on the LDH surface. Results of time-resolved DLS experiments showed that the samples were moderately stable at low polyelectrolyte concentrations. However, increasing the ionic strength gave rise to unstable dispersions in this regime and also near the IEP regardless of the salt level. Face-to-face orientation of the particles was found in TEM images recorded for the aggregated samples. Further increasing the heparin dose, the particles were restabilized where they possessed sufficiently high negative charge due to the charge reversal process. Highly stable LDH dispersions were obtained by completely covering their surface with the heparin macromolecules. Such stabilizing effect led to about 20-time increase in the CCC of the coated particles, indicating a significant resistance against aggregation even at higher electrolyte concentrations. These results allow designing efficient delivery systems where aggregation of nanocarrier LDH platelets can be tuned with biocompatible heparin macromolecules.Acknowledgements
The present research was supported by the Swiss National Science Foundation (150162), Swiss Scientific Exchange Program (SCIEX-14033), Swiss Secretariat for Education, Research and Innovation (C15.0024) as well as COST Action CM1303. The authors are grateful to Professor Michal Borkovec for providing access to the light scattering instruments in the Laboratory of Colloid and Surface Chemistry at the University of Geneva. Financial support to Z. G. was also provided by the National Health and Medical Research Council of Australia (NHMRC) Early Career Fellowship (APP1073591). We thank Professor Zhi Ping Xu for his useful comments on the manuscript.References
- B. Delalat, V. C. Sheppard, S. Rasi Ghaemi, S. Rao, C. A. Prestidge, G. McPhee, M. L. Rogers, J. F. Donoghue, V. Pillay, T. G. Johns, N. Kroger and N. H. Voelcker, Nat. Commun., 2015, 6, 8791 CrossRef CAS PubMed.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS.
- J. H. Choy, S. Y. Kwak, Y. J. Jeong and J. S. Park, Angew. Chem., Int. Ed., 2000, 39, 4042–4045 CrossRef CAS.
- V. Brunetti, L. M. Bouchet and M. C. Strumia, Nanoscale, 2015, 7, 3808–3816 RSC.
- H. F. Bao, J. P. Yang, Y. Huang, Z. P. Xu, N. Hao, Z. X. Wu, G. Q. Lu and D. Y. Zhao, Nanoscale, 2011, 3, 4069–4073 RSC.
- M. Halma, C. Mousty, C. Forano, M. Sancelme, P. Besse-Hoggan and V. Prevot, Colloids Surf., B, 2015, 126, 344–350 CrossRef CAS PubMed.
- N. K. Singh, Q. V. Nguyen, B. S. Kim and D. S. Lee, Nanoscale, 2015, 7, 3043–3054 RSC.
- Z. Gu, J. J. Atherton and Z. P. Xu, Chem. Commun., 2015, 51, 3024–3036 RSC.
- L. Perioli and C. Pagano, Expert Opin. Drug Delivery, 2012, 9, 1559–1572 CrossRef CAS PubMed.
- S. Vial, V. Prevot, F. Leroux and C. Forano, Microporous Mesoporous Mater., 2008, 107, 190–201 CrossRef CAS.
- Z. Gu, B. E. Rolfe, Z. P. Xu, J. H. Campbell, G. Q. Lu and A. C. Thomas, Adv. Healthcare Mater., 2012, 1, 669–673 CrossRef CAS PubMed.
- C. H. Zhou, Z. F. Shen, L. H. Liu and S. M. Liu, J. Mater. Chem., 2011, 21, 15132–15153 RSC.
- Z. Ferencz, A. Kukovecz, Z. Konya, P. Sipos and I. Palinko, Appl. Clay Sci., 2015, 112, 94–99 CrossRef.
- C. P. Chen, L. K. Yee, H. Gong, Y. Zhang and R. Xu, Nanoscale, 2013, 5, 4314–4320 RSC.
- M. Chen, H. M. Cooper, J. Z. Zhou, P. F. Bartlett and Z. P. Xu, J. Colloid Interface Sci., 2013, 390, 275–281 CrossRef CAS PubMed.
- H. Y. Dong, H. S. Parekh and Z. Ping, J. Colloid Interface Sci., 2015, 437, 10–16 CrossRef CAS PubMed.
- S. D. Li, J. H. Li, C. J. Wang, Q. Wang, M. Z. Cader, J. Lu, D. G. Evans, X. Duan and D. O'Hare, J. Mater. Chem. B, 2013, 1, 61–68 RSC.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- H. Hu, K. M. Xiu, S. L. Xu, W. T. Yang and F. J. Xu, Bioconjugate Chem., 2013, 24, 968–978 CrossRef CAS PubMed.
- H. Zuo, Z. Gu, H. Cooper and Z. P. Xu, J. Colloid Interface Sci., 2015, 459, 10–16 CrossRef CAS PubMed.
- M. Pavlovic, M. Adok-Sipiczki, C. Nardin, S. Pearson, E. Bourgeat-Lami, V. Prevot and I. Szilagyi, Langmuir, 2015, 31, 12609–12617 CrossRef CAS PubMed.
- E. D. E. Hyde, R. Moreno-Atanasio, P. A. Millner and F. Neville, J. Phys. Chem. B, 2015, 119, 1726–1735 CrossRef CAS PubMed.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef CAS PubMed.
- A. L. Papa, J. Boudon, V. Bellat, A. Loiseau, H. Bisht, F. Sallem, R. Chassagnon, V. Berard and N. Millot, Dalton Trans., 2015, 44, 739–746 RSC.
- B. Casu, A. Naggi and G. Torri, Carbohydr. Res., 2015, 403, 60–68 CrossRef CAS PubMed.
- H. Z. Huang and X. R. Yang, Carbohydr. Res., 2004, 339, 2627–2631 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, Biosens. Bioelectron., 2008, 23, 1285–1290 CrossRef CAS PubMed.
- R. Cao and B. X. Li, Chem. Commun., 2011, 47, 2865–2867 RSC.
- T. G. Shutava, P. P. Pattekari, K. A. Arapov, V. P. Torchilin and Y. M. Lvov, Soft Matter, 2012, 8, 9418–9427 RSC.
- J. Ramos, J. Forcada and R. Hidalgo-Alvarez, Chem. Rev., 2014, 114, 367–428 CrossRef CAS PubMed.
- A. M. E. Abdalla, L. Xiao, C. X. Ouyang and G. Yang, Nanoscale, 2014, 6, 14141–14152 RSC.
- S. R. Hwang and Y. Byun, Expert Opin. Drug Delivery, 2014, 11, 1955–1967 CrossRef CAS PubMed.
- Z. Gu, B. E. Rolfe, Z. P. Xu, A. C. Thomas, J. H. Campbell and G. Q. M. Lu, Biomaterials, 2010, 31, 5455–5462 CrossRef CAS PubMed.
- Z. Gu, A. C. Thomas, Z. P. Xu, J. H. Campbell and G. Q. Lu, Chem. Mater., 2008, 20, 3715–3722 CrossRef CAS.
- H. Zhang, Z. P. Xu, G. Q. Lu and S. C. Smith, J. Phys. Chem. C, 2010, 114, 12618–12629 CAS.
- Y. Q. Shu, P. G. Yin, B. L. Liang, S. S. Wang, L. C. Gao, H. Wang and L. Guo, J. Mater. Chem., 2012, 22, 21667–21672 RSC.
- X. D. Sun and S. K. Dey, J. Colloid Interface Sci., 2015, 458, 160–168 CrossRef CAS PubMed.
- E. Geraud, V. Prevot, J. Ghanbaja and F. Leroux, Chem. Mater., 2006, 18, 238–240 CrossRef CAS.
- D. Sranko, A. Pallagi, E. Kuzmann, S. E. Canton, M. Walczak, A. Sapi, A. Kukovecz, Z. Konya, P. Sipos and I. Palinko, Appl. Clay Sci., 2010, 48, 214–217 CrossRef CAS.
- Z. P. Xu, G. Stevenson, C. Q. Lu and G. Q. Lu, J. Phys. Chem. B, 2006, 110, 16923–16929 CrossRef CAS PubMed.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS PubMed.
- H. Holthoff, S. U. Egelhaaf, M. Borkovec, P. Schurtenberger and H. Sticher, Langmuir, 1996, 12, 5541–5549 CrossRef CAS.
- M. Owczarz, A. C. Motta, M. Morbidelli and P. Arosio, Langmuir, 2015, 31, 7590–7600 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS PubMed.
- G. Stoica, M. Santiago, S. Abello and J. Perez-Ramirez, Solid State Sci., 2010, 12, 1822–1830 CrossRef CAS.
- T. Bujdoso, A. Patzko, Z. Galbacs and I. Dekany, Appl. Clay Sci., 2009, 44, 75–82 CrossRef CAS.
- J. Hong, Z. L. Zhu, H. T. Lu and Y. L. Qiu, RSC Adv., 2014, 4, 5156–5164 RSC.
- J. Faraudo and A. Martin-Molina, Curr. Opin. Colloid Interface Sci., 2013, 18, 517–523 CrossRef CAS.
- J. Lyklema, Colloids Surf., A, 2006, 291, 3–12 CrossRef CAS.
- M. Quesada-Perez, E. Gonzalez-Tovar, A. Martin-Molina, M. Lozada-Cassou and R. Hidalgo-Alvarez, ChemPhysChem, 2003, 4, 235–248 CrossRef CAS PubMed.
- Z. P. Xu, Y. G. Jin, S. M. Liu, Z. P. Hao and G. Q. Lu, J. Colloid Interface Sci., 2008, 326, 522–529 CrossRef CAS PubMed.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- Y. Adachi, L. L. Feng and M. Kobayashi, Colloids Surf., A, 2015, 471, 38–44 CrossRef CAS.
- J. Sabin, C. Vazquez-Vazquez, G. Prieto, F. Bordi and F. Sarmiento, Langmuir, 2012, 28, 10534–10542 CrossRef CAS PubMed.
- I. Popa, G. Gillies, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2010, 114, 3170–3177 CrossRef CAS PubMed.
- C. Schneider, M. Hanisch, B. Wedel, A. Jusufi and M. Ballauff, J. Colloid Interface Sci., 2011, 358, 62–67 CrossRef CAS PubMed.
- M. Jobbagy and A. E. Regazzoni, J. Colloid Interface Sci., 2013, 393, 314–318 CrossRef CAS PubMed.
- A. Jusufi, O. Borisov and M. Ballauff, Polymer, 2013, 54, 2028–2035 CrossRef CAS.
- A. Abraham, A. Mezei and R. Meszaros, Soft Matter, 2009, 5, 3718–3726 RSC.
- D. F. Evans and H. Wennerstrom, The Colloidal Domain, John Wiley, New York, 1999 Search PubMed.
- G. Gillies, W. Lin and M. Borkovec, J. Phys. Chem. B, 2007, 111, 8626–8633 CrossRef CAS PubMed.
- T. Szabo, V. Toth, E. Horvath, L. Forro and I. Szilagyi, Langmuir, 2015, 31, 42–49 CrossRef CAS PubMed.
- L. Ghimici and M. Nichifor, Carbohydr. Polym., 2013, 98, 1637–1643 CrossRef CAS PubMed.
- D. Bauer, E. Killmann and W. Jaeger, Colloid Polym. Sci., 1998, 276, 698–708 CAS.
- S. Schwarz, S. Bratskaya, W. Jaeger and B. R. Paulke, J. Appl. Polym. Sci., 2006, 101, 3422–3429 CrossRef CAS.
- G. Fritz, V. Schadler, N. Willenbacher and N. J. Wagner, Langmuir, 2002, 18, 6381–6390 CrossRef CAS.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- Z. Y. Meng, S. M. Hashmi and M. Elimelech, J. Colloid Interface Sci., 2013, 392, 27–33 CrossRef CAS PubMed.
- J. A. Gursky, S. D. Blough, C. Luna, C. Gomez, A. N. Luevano and E. A. Gardner, J. Am. Chem. Soc., 2006, 128, 8376–8377 CrossRef CAS PubMed.
- G. P. Liu, S. Y. Liu, X. Q. Dong, F. Yang and D. J. Sun, J. Colloid Interface Sci., 2010, 345, 302–306 CrossRef CAS PubMed.
- E. Tombacz and M. Szekeres, Appl. Clay Sci., 2004, 27, 75–94 CrossRef CAS.
- L. Wu, L. Liu, B. Gao, R. Munoz-Carpena, M. Zhang, H. Chen, Z. H. Zhou and H. Wang, Langmuir, 2013, 29, 15174–15181 CrossRef CAS PubMed.
- C.-W. Chiu and J.-J. Lin, Prog. Polym. Sci., 2012, 37, 406–444 CrossRef CAS.
- M. Pavlovic, R. Huber, M. Adok-Sipiczki, C. Nardin and I. Szilagyi, Soft Matter Search PubMed , submitted.
- E. Horváth, L. Grebikova, P. Maroni, T. Szabó, A. Magrez, L. Forró and I. Szilagyi, ChemPlusChem, 2014, 79, 592–600 CrossRef.
- T. Oncsik, G. Trefalt, M. Borkovec and I. Szilagyi, Langmuir, 2015, 31, 3799–3807 CrossRef CAS PubMed.
- M. Pavlovic, M. Adok-Sipiczki, E. Horvath, T. Szabo, L. Forro and I. Szilagyi, J. Phys. Chem. C, 2015, 119, 24919–24926 CAS.
- H. G. Bagaria, K. Y. Yoon, B. M. Neilson, V. Cheng, J. H. Lee, A. J. Worthen, Z. Xue, C. Huh, S. L. Bryant, C. W. Bielawski and K. P. Johnston, Langmuir, 2013, 29, 3195–3206 CrossRef CAS PubMed.
- E. Seyrek, J. Hierrezuelo, A. Sadeghpour, I. Szilagyi and M. Borkovec, Phys. Chem. Chem. Phys., 2011, 13, 12716–12719 RSC.
- H. G. Bagaria, Z. Xue, B. M. Neilson, A. J. Worthen, K. Y. Yoon, S. Nayak, V. Cheng, J. H. Lee, C. W. Bielawski and K. P. Johnston, ACS Appl. Mater. Interfaces, 2013, 5, 3329–3339 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26072h |
| This journal is © The Royal Society of Chemistry 2016 |