
In situ calcium phosphate deposition in hydrogels of poly(acrylic acid)–polyacrylamide interpenetrating polymer networks†
M. S. Simeonov,
A. A. Apostolov and
E. D. Vassileva*
Sofia University, Faculty of Chemistry and Pharmacy, Bulgaria. E-mail: evassileva@chem.uni-sofia.bg
First published on 2nd February 2016
Abstract
Interpenetrating networks of poly(acrylic acid) and polyacrylamide were used for the first time as templates for in situ calcium phosphate (CP) deposition in an attempt to mimic the naturally occurring biomineralization. The process of CP deposition was controlled by two main factors: the functionality and the overall crosslinking density of the IPNs' gels, both factors being varied via the IPNs' composition. The approach allowed the control of CP deposition in terms of amount, type and crystallite size. In this way the IPNs' potential as a template for controlled biomineralization was demonstrated. The obtained new inorganic–organic composite materials were characterized and their further potential in the fields of bone regeneration and substitution was revealed.
1. Introduction
Biomineralization in nature is guided by anionic proteins, which regulate and control the process of mineral deposition. For example, mollusks build their shells by using a hydrophobic silk gel, aspartic acid rich proteins, and an amorphous precursor phase from which the calcium carbonate crystals form.1 Natural bone consists of a collagen I fibril matrix on which carbonated apatite is deposited. The deposition process is promoted by anionic proteins which serve as nucleating agents, the anionic groups' concentration in the guiding proteins depending on the bone's type and age.2Following the nature's lessons, the incorporation of anionic groups onto a polymer backbone became a common approach for polymer scaffolds mineralization.2,3 The crystal growth in gels has emerged as a popular platform for biomineralization processes, the interest being further enhanced by the growing number of gel-like matrices identified to play key role in the mineralization performed by biological organisms.4 The gel chemical functionality defines the way the gel interacts with solutes and thus either suppresses or enhances nucleation.5,6 On the one hand, when the gel functionality acts as heterogeneous nuclei, higher nucleation densities will be observed within the gels than in solution. On the other hand, the nucleation in gels, due to the diffusion-limited supply of reactants, takes place only when solutes accumulate to higher supersaturation than in solution. Thus besides the functionality, the crosslinking density of gels is another tool to influence the in situ crystal growth. In this way, in gels a controllable number of heterogeneous nuclei are formed but the probability for these nuclei to reach the critical size required for growth is reduced. Usually, gels yield smaller number of crystals with larger sizes compared to the solution-grown crystals.4
In general, the crystal growth in hydrogels becomes an alternative to solution-based strategies as it provides different ways to control the mineral crystallization, e.g. the rate of nucleation, the number and the size of crystals, their morphology, etc.
Calcium phosphate based materials are widely used for bone augmentation and restoration. Brushite (dicalcium phosphate dihydrate, DCPD, CaHPO4·2H2O) and monetite (dicalcium phosphate anhydrous, DCPA, CaHPO4) are of considerable interest in this respect due to their metastable nature in body fluids.7 Both exhibit the ability to be resorbed under physiological conditions and their resorptive capacity and solubility defines their osteoconductive and osteoinductive properties.8 The interest towards such kind of materials is increasing in recent years as they quickly resorb in the body and give rise to a newly regenerated bone tissue.9
Interpenetrating polymer networks (IPNs) are combination of two or more polymer networks which are not covalently bonded but penetrate into each other.10 In order to separate them one needs to break covalent bonds. This fact lies behind their very efficient energy absorbing ability which defines e.g. the IPNs excellent damping properties. When obtained via the sequential method, IPNs are characterized by a phase separated morphology where the 1st network acts as a matrix within which nodules from the 2nd one are finely dispersed (Scheme 1). When the 1st network is densely crosslinked the nodules from the 2nd network are nanosized, i.e., a nanophase separated structure could be formed. With increasing the ratio of the 2nd to the 1st network, the nodules from the 2nd network start to touch each other and the nodular morphology is transformed into an inter-connected nodular structure.10 In this way an inversing of the phases could occur. This specific and controllable by the IPNs composition only nanophase separated structure could be utilized for guiding the biomineralization processes – an approach which is novel and not explored until now.
 | ||
| Scheme 1 Schematic presentation of the IPNs of (A) PAAm/PAA and (B) PAA/PAAm. | ||
The aim of this study was to exploit the potential of IPNs as hydrogel media for crystal growth and to demonstrate that the proper selection of IPNs components (functionality) as well as the IPN composition (network density and nanostructure) are powerful tools for controlling the in situ mineralization process. This novel approach was demonstrated by utilizing the IPNs of PAA and PAAm as templates for biomimetic mineralization.
2. Experimental section
2.1. Materials
Acrylamide (AAm, purum, ≥98.0%) was purchased from Fluka AG, Germany. Acrylic acid (AA, anhydrous, 99%) was purchased from Across Organics, Belgium. Potassium peroxodisulfate (PPS) and N,N′-methylenebisacrylamide (MBAA) were purchased from Sigma-Aldrich. Potassium hydrogen phosphate, K2HPO4 (anhydrous) was purchased from Merck, Germany and calcium chloride CaCl2 (anhydrous) was purchased from Sigma-Aldrich. All reagents were used as received without further purification.2.2. IPNs synthesis
The IPNs of poly(acrylic acid) (PAA) and polyacrylamide (PAAm) were synthesized via the sequential method. Two types of IPNs were obtained, differing in the order of formation of both networks, respectively IPNs PAAm/PAA and IPNs PAA/PAAm (Scheme 1).At the next step five PAAm SNs were left to swell in acrylic acid (AA) aqueous solutions with concentrations 1 M, 2 M, 3 M, 4 M, and 5 M, respectively, containing also 0.1 wt% PPS and 4 wt% MBAA (both % relatively to AA). The in situ crosslinking polymerization of acrylic acid into SN PAAm took place at 60 °C for 5 hours. In this way five IPNs with different PAAm/PAA ratio were obtained (Table 1). Each of the newly synthesized IPNs PAAm/PAA was washed in distilled water to completely remove traces from non-reacted chemicals. The AA conversion to PAA network was determined to be 98% ± 1% by titration with NaOH of the waste waters from the washing procedure.
| Sample designation | SN PAAm | IPN PAA/PAAm | IPN PAAm/PAA | SN PAA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IPN1 | IPN2 | IPN3 | IPN4 | IPN5 | IPN6 | IPN7 | IPN8 | IPN9 | IPN10 | |||
| φPAA | 0 | 0.20 | 0.24 | 0.26 | 0.32 | 0.45 | 0.53 | 0.64 | 0.65 | 0.68 | 0.69 | 1 |
For both synthetic procedures, high concentration of the crosslinking agent MBAA (4 wt%) was used as a way to decrease the size of the 2nd network domains, i.e. to attain nano dimension of the phase separated IPNs' structure.
Following these two synthetic procedures, five PAA/PAAm and five PAAm/PAA IPNs with different ratio between both components were obtained (Table 1). For sake of comparison one SN PAA and one SN PAAm were used as referent samples.
Table 1 summarizes all prepared SNs and IPNs and each network is designated in terms of its PAA weight fraction determined by the equation:
 | (1) |
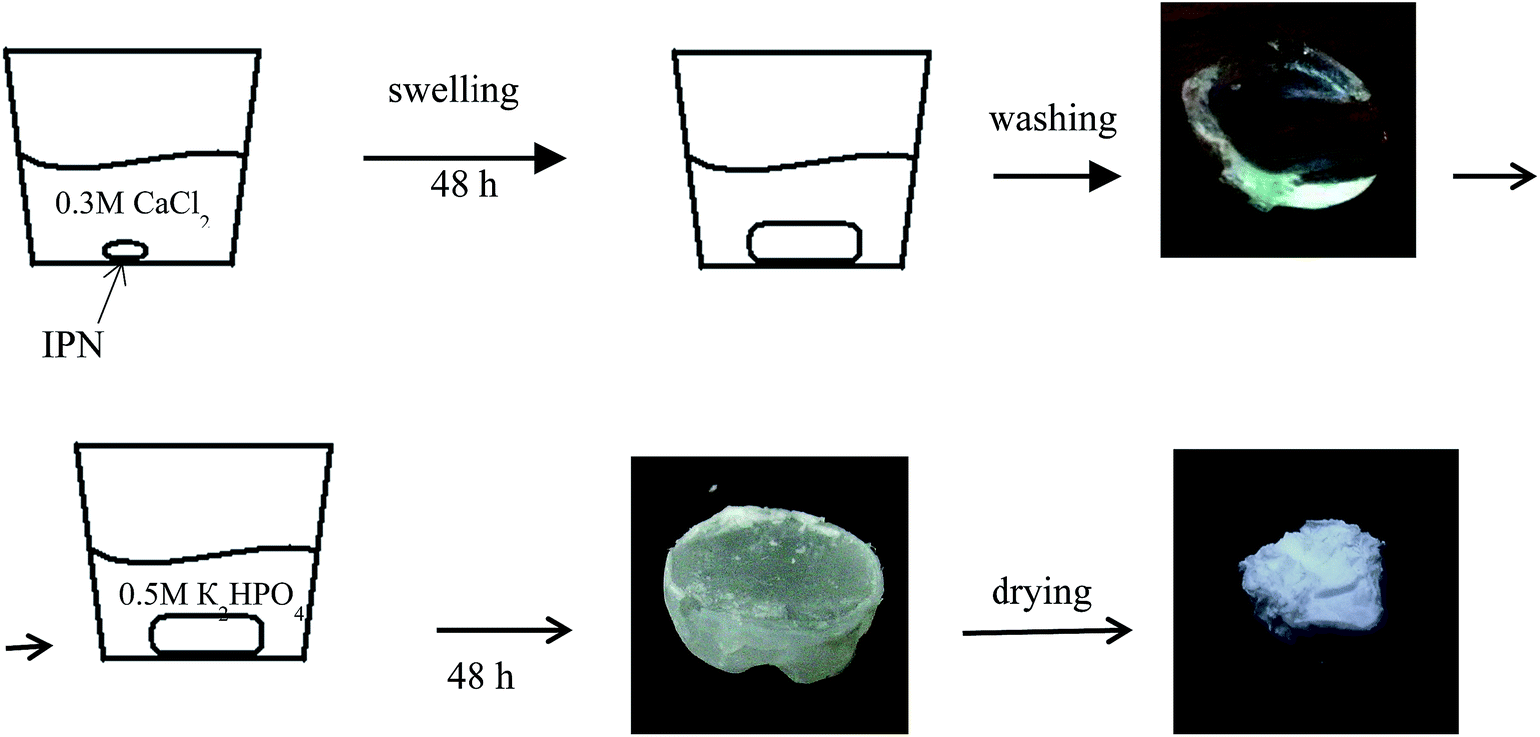 | ||
| Scheme 2 Procedure of the in situ calcium phosphates (CP) formation in IPNs. | ||
The amount of the in situ formed CP (wCP) was determined gravimetrically after washing out for 24 h in distilled water the inorganic salts (e.g. KCl), formed as side products from the CP precipitation:
 | (2) |
The successful washing was checked via scanning electron microscopy (SEM) using energy dispersive spectroscopy (EDS) analysis, the results are presented in the ESI (Fig. S1†).
2.3. IPNs' characterization
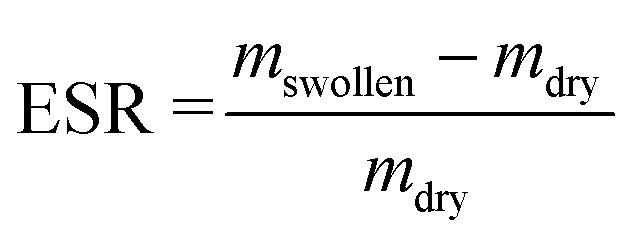 | (3) |
 | (4) |
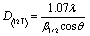 | (5) |
| β2 = B2 − b2 | (6) |
3. Results and discussion
3.1. Swelling properties
ESR dependence on PAA weight fraction (φPAA) is presented in Fig. 1 for both types of IPNs. It starts with drop of ESR at low φPAA as compared to the ESR of SN PAAm (IPN PAA/PAAm, solid symbols in Fig. 1). Similar drop at low φPAA <0.26 was already observed for IPN PAA/PAAm with similar composition in our previous work.11 It was explained by the entanglement of PAA and PAAm networks in the IPNs and as these entanglements act as physical junctions, it is expected that the ESR of the IPNs would decrease when compared to the neat PAAm.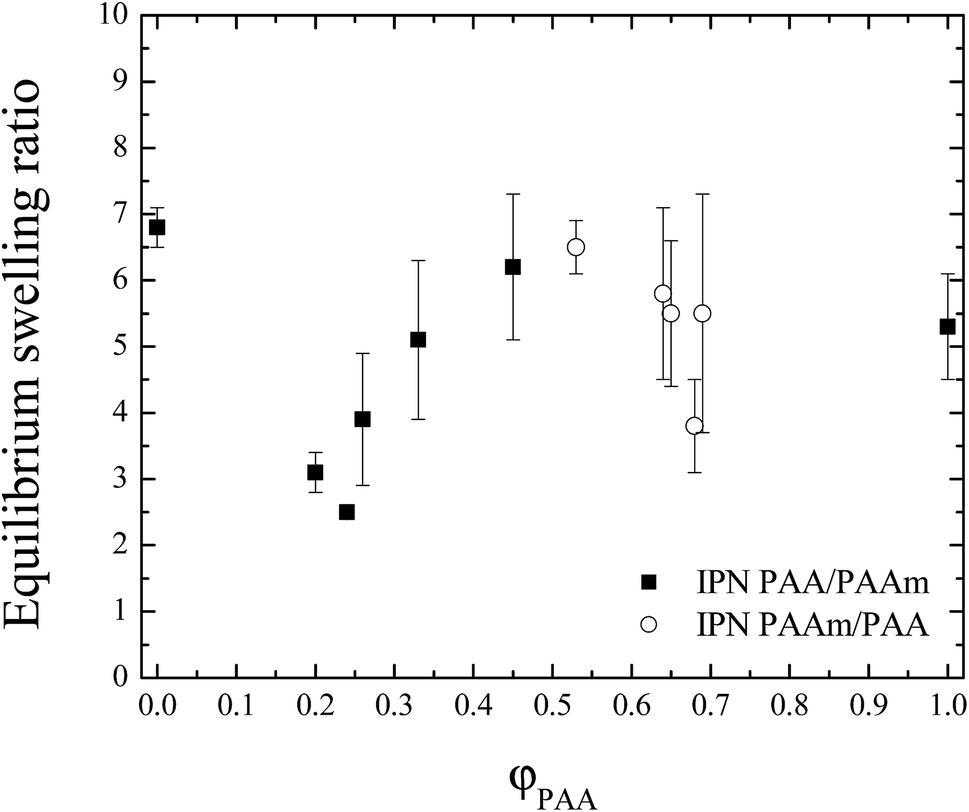 | ||
| Fig. 1 Equilibrium swelling ratios for both types of IPNs vs. IPN composition. | ||
The further PAA weight ratio increase (φPAA > 0.26) results into a IPNs' ESR increase which tends to the ESR of the neat PAA (Fig. 1). This behaviour is explained by the PAA ionization, which is high in distilled water (pKPAAa = 4.5). For example at pH = 7 about 70% of the PAA carboxylic groups are ionized.16 Thus, in the IPNs swollen state most of the acid groups in PAA are transformed into carboxylic anions.17 It is known that when PAA and PAAm are mixed to form an interpolymer complex, in fact PAA dictates the configuration of the complex.18 When φPAA increases, the contribution of the PAA to the overall behavior of the IPNs also increases. Due to the repulsion between COO−, the PAA network is stretched and hence the IPNs' ESR increases. As a result, at high PAA content, the IPNs' ESRs approach the ESR of SN PAA network.
In summary, when the PAA content in the IPNs increases, the repulsive interactions also increase due to the higher number of carboxylic anions that results into more extended PAA network, respectively into higher swelling ability of the IPNs and higher ESR values.
At lower PAA content (φPAA < 0.26), although the PAA carboxylic groups are ionized, their amount is too low to influence significantly the behavior of the whole IPN. The repulsive interactions between carboxylic anions of PAA are shielded by the PAAm chains which significantly prevail in number. Thus at low φPAA (IPN1 and IPN2), the ESR dependence on IPN composition is guided not by the PAA ionization but rather by the entanglements between PAA and PAAm chains.
One should mention here, that PAA and PAAm could form H-bonds between themselves when PAA is at low ionization degree (pH < 4.5). Then PAA could form either intramolecular or intermolecular H-bonds with itself as well as with PAAm. This happens also in solid state where the interpolymer complexation between PAA and PAAm is very strong.18 At neutral pH, the H-bonding could also occur but is less probable due to the PAA ionization.
3.2. Microhardness (MH)
In Fig. 2 the MH values of the IPNs are presented as a function of φPAA as well as the MH of both SNs PAA and PAAm. There is no clear trend for the MH dependence on the IPNs' composition. It should be however pointed out that the MH values of IPNs are higher as compared to the MH of both SNs as well as higher to the predicted values from the additivity law (solid line in Fig. 2). This confirms the mutual interlacing, i.e. the formation of additional physical junctions, between both networks PAA and PAAm in IPNs assumed above (Fig. 1). This positive deviation from the additive law reflects also the already mentioned strong propensity for interpolymer complexation and H bonding between PAA and PAAm in solid state where MH was measured.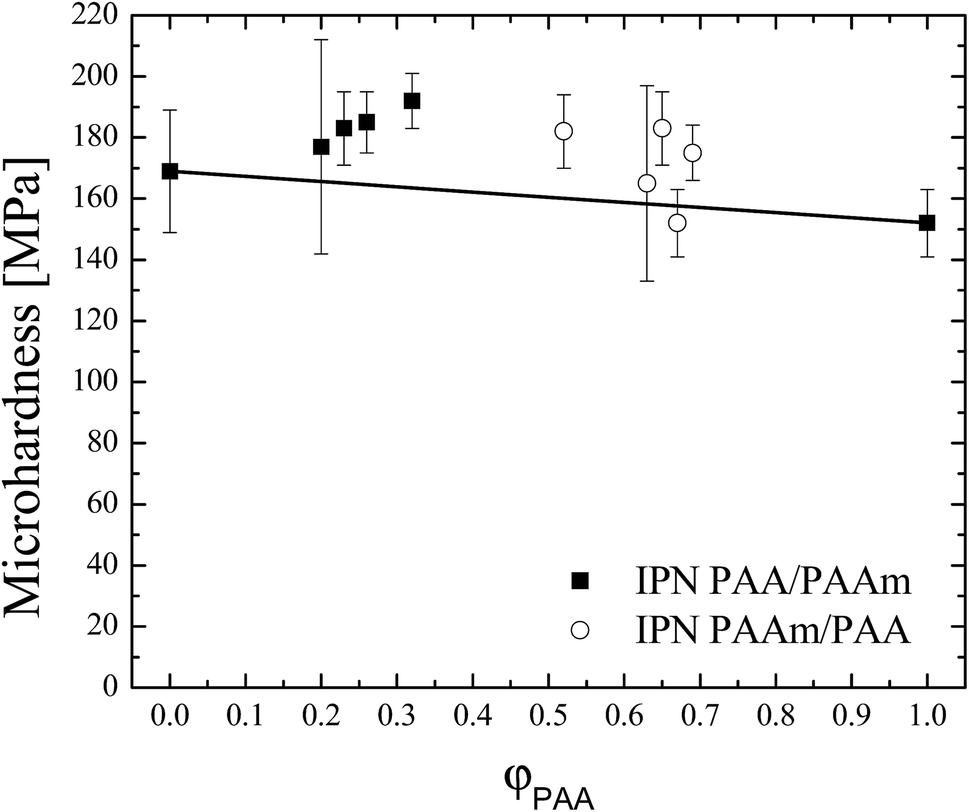 | ||
| Fig. 2 Microhardness vs. IPNs' composition. Solid line is drawn according to the additivity law. | ||
3.3. Thermal properties of IPNs
In Fig. 3 the DSC thermograms of IPNs PAAm/PAA as well as for both SNs PAA and PAAm are presented. For SN PAAm ((φPAA = 0, Fig. 3) an endothermic peak at ∼220 °C appears which is known to be due to the 1st stage of PAAm thermal decomposition. It takes place in the range of 220 to 330 °C and consists in reaction of imidization between the PAAm amino groups which results into an NH3 release.19 SN PAA is also known to have a decomposition peak at 230 °C, but in the neat PAA thermogram it could not be seen (φPAA = 1, Fig. 3). | ||
| Fig. 3 DSC thermograms of IPNs PAAm/PAA with different compositions. | ||
The onset of the PAAm degradation peak is observed for all IPNs compositions. When compared to the neat PAAm, this onset is shifted to higher temperatures, which means that the IPN formation increases the PAAm thermal stability. This thermal stability improvement of IPNs could be explained by the shielding effect that the PAA chains have on the PAAm chains, separating the amino groups and not allowing them to take part in the imidization reaction. This explanation is supported by the fact that the peak corresponding to the 1st stage of the thermal degradation of PAAm in IPNs shifts to lower temperatures as PAAm content increases. That means that as the decomposing component (PAAm) content increases, more and more PAAm chains are coming closer to each other, thus enhancing the imidization reaction. In summary, the IPNs of PAA and PAAm possess higher thermal stability when compared to the neat PAAm and their thermal stability is composition dependent.
In Fig. 3, it could be also seen that all IPNs show one very broad glass transition which lies between the Tgs of both SNs PAA and PAAm. The same was observed for the IPNs PAA/PAAm, their thermograms not presented here. One broader Tg region between the Tgs of both neat components, i.e. glass-transition temperatures of the two polymers shifting inward, is usually observed in IPNs due to an increase in the miscibility of the two polymers caused by the presence of cross-links.10
3.4. Calcium phosphates in situ formation in IPNs
The in situ precipitation of CP into the IPNs was followed gravimetrically. The weight percent of deposited CP into IPNs is shown as a function of the IPNs composition in Fig. 4. There is a clear trend of increasing the CP content as φPAA increases. This is in line with the recent findings of Yokoi et al. that an increase in the number and density of COOH groups results into a linear increase in the quantity of adsorbed calcium ions.6 | ||
| Fig. 4 Weight% of CP in situ deposited in IPNs as a function of IPNs composition. Solid line – additivity law, dashed line – linear fit (see text). | ||
In the same study, however, the authors observed the opposite trend concerning the in situ deposited CP amount in PAA and PAAm copolymers. Namely, the increase in the COOH groups number resulted into a decrease in the amount of the precipitated CP. This fact was explained by the chelation of calcium ions by the COOH groups which decreased the calcium ions activity and hence the amount of precipitated CP.6
One should mention here two main differences between our study and the one of Yokoi et al.6 Generally we have used (i) much higher (∼4 times) concentration of the crosslinking agent which imposed a nano dimension of the phase separated IPN structure (Scheme 1)11 and (ii) our samples were ∼10 times thinner as compared to the used by them. These two factors change the ions diffusion, enhancing it most probably, as compared to the copolymer system studied by Yokoi et al.6 As a result, the CP precipitation changed although some their conclusions are valid also for our system. For example, we have also detected the influence of other than the purely concentration dependence of AA content on the CP precipitation. The black line in Fig. 4 is drawn according to the additivity law: wt% CaPO4 = 29 + 29φPAA. It is seen there that when PAA played the role of a matrix (Scheme 1, IPN PAA/PAAm) the amount of the in situ deposited CPs deviates positively from the additivity law (closed symbols in Fig. 4), tending towards the amount deposited in the neat PAA. This positive deviation from the additive law means that the CPs deposition is not simply proportional to the acidic groups' concentration in the IPN gels but is enhanced due to other factors. This result is in agreement with the expectations that not only the gel functionality governs the in situ CPs deposition in IPNs gels but also their crosslinking density as this is the factor that defines e.g. ions diffusion. Indeed when compare the CP deposition results (Fig. 4) with the ESR variation with PAA content one can see that the amount of deposited CP in fact increases with ESR increase (Fig. 1), i.e., with the enhancement of the ions diffusion in the IPNs hydrogels (IPN PAA/PAAm, closed symbols in both figures).
In contrast, when PAA played the role of the 2nd network (i.e. nodules were formed from PAA, Scheme 1, IPN PAAm/PAA), the amount of in situ deposited CPs in these IPNs deviates negatively, i.e., lower amount of CPs is deposited in the IPN than the predicted from the additivity law. This result also confirms the conclusion that the CP deposition is not simply governed by the acidic functionality availability but also other factors have influence on the process, one possibility being e.g. the changed activity of calcium ions as stated by Yokoi et al.6
The amount of the in situ deposited CP in the SN PAAm is significantly lower than the amount deposited in the SN PAA which was expected due to the PAA functionality besides the good swelling ability of both SNs. This result was also proved by X-ray measurements (see Section 3.9).
The dashed line in Fig. 4 is the linear fit drawn according to the experimental points (with error bars as weights). The regression coefficient is 0.908 and the slope is 25 ± 3, which means that both lines – the additivity law and the linear fit in fact coincide within the experimental error.
3.5. Morphology of IPNs with in situ deposited CP
The morphology of broken surfaces of IPN PAA/PAAm (IPN3, φPAA = 0.26) and IPN PAAm/PAA (IPN9, φPAA = 0.68) containing in situ formed CP were studied by SEM (Fig. 5). The morphology of the neat IPNs was already studied in our previous paper11 and their nano phase separation was detected, the size of the nodules formed from the 2nd polymer network being below 100 nm for all studied IPNs. Coming back to the current study, when PAA formed the matrix and the nodules inside were from PAAm (Scheme 1, IPN PAA/PAAm), the crystallization of CP depended on the remoteness from the edge of the sample, i.e. from the depth, similarly to the observed by Yokoi et al. for the CP crystallization in neat PAAm gels.5 At areas close to the sample edge (depth < 30 μm), all studied samples showed small fine crystals from CP formed within the matrix (Fig. 5, IPN3). At this depth, the CP crystals are formed under high supersaturation conditions due to the fact that close to the edge more ions could diffuse into the sample.5 Under these conditions, the nucleation rate is much higher thus prevailing the process of crystal growth.5 According to the same authors, these small crystals are further redissolved thus giving rise to larger ones. However, we did not observed it, most probably because we used shorter time for CP maturation – 2 days versus 5 days used in the study of Yokoi et al.5 | ||
| Fig. 5 Morphology of broken surfaces of both types IPNs with in situ precipitated CP. | ||
At larger distance from the sample edge (depth > 30 μm) for the same IPN PAA/PAAm, the observed CP crystals are larger due to the fact that in the core of the sample the ions are in a lower saturation level which promotes the crystal growth rather than the nucleation. This was the case observed for neat PAAm gel with in situ deposited CP.5
When PAAm was the matrix and PAA nodules were dispersed within (Scheme 1, IPN PAAm/PAA), the sample morphology appeared not to depend on the sample thickness and all over the broken surface the small CP grains could be seen (Fig. 5, IPN 9). The above mentioned ∼4 times higher concentration of the crosslinking agent as compared to the studies of Ohtsuki group5,6 most probably defined the much smaller size of the CP crystals observed for both types of IPNs in the current study.
The small CP grain size, observed for all IPNs samples with in situ formed CP, was further proved by TEM, performed on the ashes left after removing the organic part of the sample after heating up to 900 °C (Fig. S2, ESI†). There, the nanometer size of the CP grains could be seen, confirming the small size of the in situ formed CP within the IPNs matrices. Thus, the influence of the IPNs network density and structure on the CPs formation, i.e. on biomineralization process was confirmed.
3.6. Thermal properties of IPNs with in situ deposited CPs
The in situ CP deposition in the IPNs should affect the latters' thermal properties as it results in the formation of organic–inorganic composite materials. As it could be seen in Fig. 6, Tg of the IPNs increases after the in situ CP deposition which is in line with the observed Tg increase when inorganic–organic composite material is formed.20 The Tg increase in organic–inorganic polymer composites is due to the partial immobilization of the polymer chains around the inorganic filler particles. The stronger the interaction between the inorganic particles and the polymer matrix is, the higher is the Tg increase of the composite material. Transferred to the current system, that means that the CP particles interact with the polymer chains in their vicinity thus immobilizing them. As a result, the Tg of the polymer matrix increases. In Fig. 5 the thermograms of two IPNs with different compositions are presented without and with in situ deposited CP. For both compositions an increase in the polymer matrix Tg is observed which confirms the assumed interaction between the deposited CP and the polymer matrices. | ||
Fig. 6 DSC thermograms of IPNs PAAm/PAA with (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) and without ( ) and without (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) in situ deposited CP. The IPNs compositions are designated in the figure. ) in situ deposited CP. The IPNs compositions are designated in the figure. | ||
The in situ deposited CP influence also the IPNs degradation temperatures. As it could be seen in Fig. 6, the CP deposition decreases the IPNs' degradation temperature and widens the degradation peak. For comparison, the thermogram of the neat PAAm is presented where the degradation peak around ∼220 °C is clearly seen. As the amount of deposited CP in the IPNs increases (it increases with φPAA, as shown in Fig. 4), the degradation peak shifts to lower temperatures and becomes wider.
It is shown in literature, that when amorphous calcium phosphates (ACP) and hydroxyapatites (HAP) are deposited in neat PAAm, the PAAm degradation onset is not influenced by the CP presence,21,22 i.e., the imidization stage in the PAAm degradation starts as normal at ∼220 °C. However, for PAA based materials, the presence of HAP causes decrease of the PAA degradation onset,23 which could explain the observed here decrease in the IPNs' degradation temperature and widening of the degradation peak.
Similar effect on PAA degradation has also Cu(NO3)2 where as the Cu content increases, the PAA degradation peak shifts from 250 °C to almost 150 °C.24 Thus the presence of CP influences the thermal stability of the IPNs via decreasing the PAA degradation temperature.
3.7. IR characterization of in situ deposited CPs into SNs of PAA and PAAm
In Fig. 7A, the IR spectra of the neat SN PAAm as well as of the SN PAAm with in situ deposited CP (designated as PAAm+CP) are presented. Both spectra are quite similar as they both contain the typical for PAAm bands24,25 (see ESI, Table S1†). | ||
| Fig. 7 AT-FTIR spectra of: (A) neat PAAm and PAAm with in situ deposited CP (PAAm+CP) and (B) neat PAA and PAA with in situ deposited CP (PAA+CP). | ||
In the PAAm+CP the typical amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at ∼1655 cm−1 is slightly shifted (∼1653 cm−1) to lower frequency (resolution of the spectra is ∼2 cm−1) which means that this group possibly interacts with the in situ deposited CP. The low frequency shift of the CO stretching vibration in amides was shown to indicate that the CO bonds in the peptide chain were weakened because of the formation of new chelate bonds between Ca2+ and CO bonds.26
O stretch at ∼1655 cm−1 is slightly shifted (∼1653 cm−1) to lower frequency (resolution of the spectra is ∼2 cm−1) which means that this group possibly interacts with the in situ deposited CP. The low frequency shift of the CO stretching vibration in amides was shown to indicate that the CO bonds in the peptide chain were weakened because of the formation of new chelate bonds between Ca2+ and CO bonds.26
When comparing PAAm and PAAm+CP spectra two peaks appear in the latter which are not seen in the former; these are denoted in bold and with asterisk in Fig. 7A and are explained in Table 2.
| Band position | Attribution | Remark |
|---|---|---|
| ∼1118 cm−1 | Antisymmetric stretching  in CP in CP |
This band usually appears in 900–1200 cm−1, however as the band is neither split nor there is a shoulder, it corresponds to amorphous CP, which coincides with the X-ray data (see Section 3.9) |
| ∼3732 cm−1 | Free OH stretching | Since this peak does not appear in the neat PAAm spectrum, it originates from the in situ deposited CP |
The IR spectra of SN PAA (PAA) and PAA with in situ deposited CP (designated as PAA+CP) again have quite many common bands24 (see ESI, Table S2†). The new bands that appear in the PAA+CP sample as compared to the neat PAA are in bold and marked with asterisk in Fig. 7B and are summarized in Table 3.
| Band position | Attribution | Remark |
|---|---|---|
| ∼898 cm−1 | P–O–H symmetric stretching mode | This is typical for acidic CP, i.e. this is the most characteristic feature of the presence of HPO42− species (ref. 28) |
| ∼1082 cm−1 |  stretching stretching |
Coming from the in situ precipitated CP |
| ∼1558 cm−1 | Asymmetric COO− stretching  of the carboxylic anion (ref. 27) of the carboxylic anion (ref. 27) |
Means that COOH groups in PAA+CP are transformed into COO− |
| ∼1635 cm−1 | Stretching asymmetric vibration of the carboxylate group in PAA salts (ref. 26) | Means that COOH groups in PAA+CP are transformed into COO− as the stretching of CO from COOH disappears in the PAA+CP |
| ∼3732 cm−1 | Free OH stretching from the in situ deposited CPs | Similarly to the case of PAAm+CP, as it was explained in Table 2 |
The transformation of carboxylic group to carboxylic anion is expected as the carboxylic groups in PAA are assumed to interact with Ca2+ and in this way to initiate the CP deposition into the hydrogels. Another proof for this transition is the doublet at 1161 cm−1 and 1236 cm−1 (for νC–O + δO–H) which appears only in the neat PAA and disappears in the PAA+CP because of the formation of carboxylic anion.
In contrast to PAAm+CP spectrum where the band at ∼2930 cm−1 was assigned to the C–H stretching, in the PAA+CP IR spectrum this band is obscured by the broader OH stretching.
In the spectra of PAAm+CP and PAA+CP two bands appear which are due to CO2 from air, namely a weak band at 667 cm−1 as well as two bands at 2339 cm−1 and 2359 cm−1 coming from the CO2 in the beam.
In summary, the CP deposited in both SNs give a typical broad phosphate band due to stretching  modes in the 1000–1100 cm−1 region with two distinctive peaks at ∼1016 cm−1 in PAAm+CP spectrum and at ∼1082 cm−1 in the PAA+CP spectrum. Moreover, in the PAA+CP IR spectrum the typical band for HPO42− groups appears which means that the neat PAA promotes the formation of acidic CPs. In the PAAm+CP IR spectrum this band is obscured by the end of the spectrum.
modes in the 1000–1100 cm−1 region with two distinctive peaks at ∼1016 cm−1 in PAAm+CP spectrum and at ∼1082 cm−1 in the PAA+CP spectrum. Moreover, in the PAA+CP IR spectrum the typical band for HPO42− groups appears which means that the neat PAA promotes the formation of acidic CPs. In the PAAm+CP IR spectrum this band is obscured by the end of the spectrum.
3.8. IR characterization of the in situ deposited CP in IPNs of PAA and PAAm
In Fig. 8 the IR spectra of IPNs+CP (PAAM+CP and PAA+CP) for several different IPNs' compositions are presented. Several main bands coming from amide or carboxyl groups could be seen there along with the newly appeared bands coming from the in situ formed CP. Table 4 summarizes the new bands in the IPN+CP samples coming from the CP or appearing due to the CP formation. | ||
| Fig. 8 AT-FTIR spectra of IPNs of PAA and PAAm with in situ deposited CP. | ||
| Band position | Attribution | Remark |
|---|---|---|
| ∼3000–3600 cm−1 | Broad band due to the OH stretching from the deposited CPs | Appears in all IPN+CP samples (designated with a straight line) |
| ∼1652 cm−1 | CO stretching vibration in carboxylic anion |
Seen in all samples IPN+CP for IPN with φPAA >0.5 |
| ∼1016 cm−1 and ∼1082 cm−1 | Stretching  modes modes |
Typical broad phosphate band in the 1000–1200 cm−1 region with two distinctive peaks |
| ∼898 cm−1 | P–O–H symmetric stretching mode | The band was observed in the neat PAA+CP and confirms the formation of HPO42− groups when in situ CP were deposited into the IPN hydrogels |
As the bands due to PO4 are very broad for all IPNs compositions that means that the precipitated CP in all samples are poorly crystalline which coincides with the results for both SNs with in situ deposited CP: PAA+CP and PAAm+CP. This data are also confirmed by the X-ray study (see Section 3.9).
3.9. X-ray characterization of in situ deposited CPs into SNs and IPNs of PAA and PAAm
In Fig. 9 the X-ray diffractograms of three samples – both neat SNs and the IPN5 (φPAA = 0.45) all of them with in situ deposited CP are presented. According to the data all samples have relatively low crystallinity which coincides with the broad bands for phosphate group observed by IR. Although the CPs are poorly crystalline, three crystalline phases were identified by the search-match program Match2, namely brushite, sylvite and monetite. A semi-quantitive analysis was performed on the basis of the corundum number and the obtained results are presented in Table 5. | ||
| Fig. 9 X-ray diffractograms of the materials, obtained after in situ deposition of CP into SN PAAm, SN PAA and IPN5 (φPAA = 0.45). | ||
| Sample designation | Brushite [wt%] CaHPO4·2H2O (96-900-7306)b | D〈12![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 〉, [Å] 〉, [Å] |
Sylvite [wt%] KCl (96-900-3130) | Monetite [wt%] CaHPO4 (96-900-7620) | D〈03〉, [Å] |
|---|---|---|---|---|---|
| a Crystallite size D〈12〉 and D〈03〉 for brushite and sylvite in direction perpendicular to the (12) and (03) plane respectively.b “Crystallography Open Database” # of each phase. |
|||||
| PAAm | — | — | 76.6 | 23.4 | 360 ± 30 |
| IPN5 | 69.7 | 900 ± 27 | 30.3 | — | — |
| PAA | 89.7 | 340 ± 30 | 10.3 | — | — |
In all three samples sylvite (KCl) was identified which is expected as it is the side product of CP deposition. In the neat PAA only brushite was formed most probably due to the acidic environment ensured by the polyanion network (PAA) which promoted the brushite formation. This result coincides with the IR data about HPO42− species presence as well as with the COOH transformation to COO− in the same sample (see Section 3.7).
In the neat PAAm only monetite was formed. After comparison of both diffractograms, it appears that by following the same procedure and solution concentrations for CP in situ deposition, more CP was deposited in the neat SN PAA than in the neat SN PAAm. This is in agreement with the gravimetrically obtained data about CP deposition in both samples (Fig. 4).
In the IPN5 (φPAA = 0.45) only brushite phase of the CP was formed and this could be explained by the PAA leading role in the CP deposition process. This also coincides with the IR data where for all IPN+CP materials the band for HPO42− appears.
In Table 5 the crystallite size of brushite and monetite in direction, perpendicular to the planes (12) and (03), respectively, is also presented. In order to determine them, the (12) reflections of brushite and 03 of magnetite were used since these maxima are relatively well expressed. Crystallite sizes in other directions of brushite and monetite in PAA and IPN5 were not calculated due to the lack of appropriate peaks. The crystallite size of sylvite is not discussed as it is a side product and of no interest here.
The size of the brushite crystallites in IPN5 is much higher than that in SN PAA. This could be due to the lower number of acidic groups serving as crystallization nuclei per unit volume in the former as compared to the latter. As a result, smaller number of crystals with larger sizes are formed in the IPNs as compared to the neat PAA.
The formation of brushite and respectively monetite into the studied SNs and IPNs defines their potential application as bone restoration materials. Brushite and monetite being acidic forms of calcium phosphates are relatively soluble in biological solutions, although in different pH ranges. Brushite when in contact with biological fluids tends to dissolve and re-precipitate into a less soluble calcium phosphate (e.g. hydroxyapatite)29 while monetite when implanted in vivo preserves its chemical composition and degradability, allowing replacement by the newly formed bone tissue.30 Taking in mind the poor functionality of the damaged tissue surrounding the bone defects, biomaterials based on brushite and monetite are of special interest as they can enhance the process of bone augmentation and restoration.
4. Conclusions
IPNs of PAA and PAAm have been synthesized and characterized in terms of their swelling ability, microhardness and thermal properties. For the first time these IPNs were applied as a polymer template for biomineralization taking advantage of their specific characteristics. Two key factors appear to control the in situ CP deposition in the IPNs, namely: the IPNs' functionality as well as the IPNs' hydrogels network density both controlled through variation of the IPNs' composition. Thus the IPNs' potential as a biominerazation template was successfully demonstrated.Acknowledgements
The work was financially supported by the Bulgarian National Science Fund under Contract DFNI-T02/5.References
- J. D. Addadi, F. Nudelman and S. Weiner, Chem.–Eur. J., 2006, 12, 980–987 CrossRef PubMed.
- J. D. Kretlow and A. G. Mikos, Tissue Eng., 2007, 13(5), 927–938 CrossRef CAS PubMed.
- G. Liu, D. Zhao, A. P. Tomsia, A. M. Minor, X. Song and E. Saiz, J. Am. Chem. Soc., 2009, 131, 9937–9939 CrossRef CAS PubMed.
- E. Asenath-Smith, H. Li, C. Keene, Z. W. She and L. A. Estroff, Adv. Funct. Mater., 2012, 22, 2891–2914 CrossRef CAS.
- T. Yokoi, M. Kawashita, K. Kikuta and C. Ohtsuki, Mater. Sci. Eng., C, 2010, 30, 154–159 CrossRef CAS.
- T. Yokoi, M. Kawashita and C. Ohtsuki, Journal of Asian Ceramic Societies, 2013, 1, 155–162 CrossRef.
- G. Cama, B. Gharibi, J. C. Knowles, S. Romeed, L. DiSilvio and S. Deb, J. R. Soc., Interface, 2014, 11, 20140727 CrossRef CAS PubMed.
- V. B. Idowu, G. Cama, S. Deband and L. Di Silvio, In vitro osteoinductive potential of porous monetite for bone tissue engineering, J. Tissue Eng., 2014, 5, 1–14 Search PubMed.
- T. R. Desai, S. B. Bhaduri and A. Cuneit Tas, A self-setting, monetite (CaHPO4) cement for skeletal repair, Chapter 6 in Advances in Bioceramics and Biocomposites II, Ceram. Eng. Sci. Proc., 2008, 27(6) Search PubMed.
- L. H. Sperling, in Interpenetrating polymer networks, ed. D. Klempner, L. H. Sperling and L. A. Utracki, Advances in Chemistry, American Chemical Society, Washington, DC, 1994, p. 24 Search PubMed.
- M. Simeonov, B. Kostova and E. Vassileva, Macromol. Symp., 2015, 358(1), 225–231 CrossRef CAS.
- M. Muchtardi, J. Levita, D. Rahaya and H. Rahmi, Food Publ. Health, 2012, 2(2), 16–20 CrossRef.
- MATCH!, Version 2.x, CRYSTAL IMPACT, Kreuzherrenstr. 102, 53227 Bonn, Germany, http://www.crystalimpact.com/match Search PubMed.
- H. P. Klug and L. E. Alexander, X-ray diffraction procedures for polycrystalline and amorphous materials, John Wiley & Sons, New York, 1974, p. 511 Search PubMed.
- B. D. Cullity and S. R. Stock, Elements of x-ray diffraction, Prentice Hall, New Jersey, Third edn, 2001, p. 400 Search PubMed.
- M. Edwards, M. M. Benjamin and J. N. Ryan, Colloids Surf., A, 1996, 107, 297–307 CrossRef CAS.
- M. Jiang, M. Li, M. Xiang and H. Zhou, Adv. Polym. Sci., 1999, 146, 121–196 CrossRef CAS.
- F. O. Garces, K. Sivadasan, P. Somasundaran and N. J. Turro, Macromolecules, 1994, 7, 272–278 CrossRef.
- W. M. Leung, D. E. Axelson and J. D. Van Dyke, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1825–1846 CrossRef CAS.
- D. H. Droste and A. T. Dibenedetto, J. Appl. Polym. Sci., 2003, 13(10), 2149–2168 CrossRef.
- Q.-L. Tang, K.-W. Wang, Y.-J. Zhu and F. Chen, Mater. Lett., 2009, 63(15), 1332–1334 CrossRef CAS.
- G. S. Sailaja, S. Velayudhan, M. C. Sunny, K. Sreenivasan, H. K. Varma and P. Ramesh, J. Mater. Sci., 2003, 38, 3653–3662 CrossRef CAS.
- S. Dubinsky, G. S. Grader, G. E. Shter and M. S. Silverstein, Polym. Degrad. Stab., 2004, 86, 171–178 CrossRef CAS.
- Infrared spectroscopy of polymers, ed. N. A. Del Fanti, Thermo Fisher Scientific Inc., 2008, p. 204 Search PubMed.
- W. Zhang, Z.-L. Huang, S.-S. Liao and F.-Z. Cui, J. Am. Ceram. Soc., 2003, 86, 1052–1054 CrossRef CAS.
- B. Grabowska, M. Sitarz, E. Olejnik, K. Kaczmarska and B. Tyliszczak, Spectrochim. Acta, Part A, 2015, 151, 27–33 CrossRef CAS PubMed.
- M. B. Povea, W. A. Monal, J. V. C. Rodríguez, A. M. Pat, N. B. Rivero and C. P. Covas, Mater. Sci. Appl., 2011, 2, 509–520 CAS.
- C. Holt, M. J. J. M. Van Kemenade, J. E. Harries, L. S. Nelson, R. T. Bailey, D. W. L. Hukins, S. S. Hasnain and P. L. De Bruyn, J. Cryst. Growth, 1988, 92, 239–252 CrossRef CAS.
- M. Kumar, J. Xie, K. Chittur and C. Riley, Biomaterials, 1999, 20, 1389–1399 CrossRef CAS PubMed.
- B. Idowu, G. Cama, S. Deband and L. Di Silvio, J. Tissue Eng., 2014, 5, 1–14 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26066c |
| This journal is © The Royal Society of Chemistry 2016 |