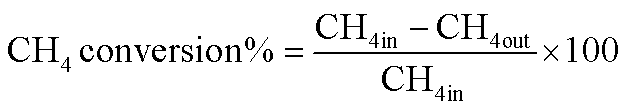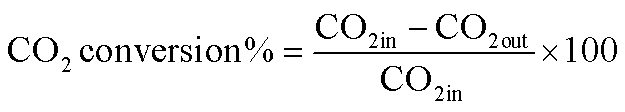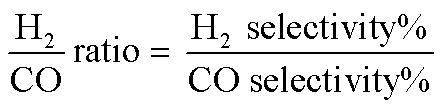DOI:
10.1039/C5RA25869C
(Paper)
RSC Adv., 2016,
6, 10372-10384
Investigation of Ce(III) promoter effects on the tri-metallic Pt, Pd, Ni/MgO catalyst in dry-reforming of methane
Received
4th December 2015
, Accepted 14th January 2016
First published on 19th January 2016
Abstract
A mixture of cerium oxide and magnesium oxide supports with certain mole ratios of Mg2+/Ce3+ were prepared via the co-precipitation of Mg and Ce nitrates, and followed by impregnation with 1 wt% each of Ni, Pd, and Pt metals to form Pt, Pd, Ni/Mg1−xCexO catalysts. Evaluation of the prepared catalysts was carried out by a DRM reaction for 200 h and they were characterised by means of in situ XRD, XRF, XPS, BET, H2-TPR, TEM and TGA. It was found that the interaction of a suitable amount of MgO with Ce2O3 stabilised a cubic phase in the catalysts, which has a high basicity to adsorb CO2 forming a monoclinic Ce2O2CO3 species in the DRM reaction. The introduction of MgO also created surface oxygen ions. The oxidisation and the removal of the deposited carbon maybe achieved by both monoclinic Ce2O2CO3 and surface oxygen, keeping the metal Ni, Pd, and Pt catalyst at high activity and stability. The Ce2O3 as a promoter in the catalyst had several effects such as: stabilisation of the magnesia cubic phase; increase in its thermal stability, increase in the basicity of the support, decrease in the carbon deposition, and decrease in the reducibility of the Ni2+, Pd2+, and Pt2+ ions.
1. Introduction
The demand for alternative energy resources has recently been on the rise due to the fast depletion of fossil fuel. Therefore, the utilisation of greenhouse gases, such as methane and carbon dioxide, as alternative energy has received much attention. It has been found that the reserves of methane are larger than crude oil reserves because methane can be produced from various sources, including shale gas, fermented wastes, and methane hydrates.1 Methane can be transformed into synthesis gas (a mixture of H2 and carbon CO, also known as syngas) through reforming reactions using steam, partial oxidation or carbon dioxide.
The synthesis gas is an important feedstock for fuels and/or the production of chemicals in industries. Coal, petroleum, natural gas, and biomass can be converted into syngas. However, varying the H2/CO molar ratios is required in accordance with the industrial applications of syngas. For example, the H2/CO ratio of 2 is required for methanol synthesis;2 whilst the ratio is controlled at 1 for dimethyl ether (DME) synthesis under the single step process.3 In the Fisher–Tropsch process, the H2/CO ratio is usually in the range of 1 to 2, depending on the type of fuel synthesized.4 Consequently, the H2/CO ratio in syngas plays an important role in fuel production. Recently, a number of studies5 that have been carried out have focused on the indirect reduction of iron oxides using H2 and CO as reducing agents. The indirect reduction of iron oxides involves two mechanisms: CO-based and H2-based.6 The two gases or syngas can be produced from the reforming of coke oven gas (COG). In this aspect, there is no limit of the H2/CO ratio in the utilisation of syngas for the iron making processes, whereby H2 and CO are able to individually trigger the indirect reduction in a blast furnace.
The conversion of CH4 to the synthesis gas, which constitutes the feed for the Fischer–Tropsch syntheses, is very important from an industrial point of view. The dominant commercial method employed to produce synthesis gas is the steam reforming of methane (eqn (1)).7
| | |
CH4 + H2O → CO + 3H2 ΔH°298K = 225.0 kJ mol−1
| (1) |
However, there are several limitations to this reaction. First, the energy consumption for this reaction is higher. Next, the selectivity for CO for this reaction is poor. Finally, the reaction is unsuitable for Fischer–Tropsch syntheses because its H2/CO ratio is high. As a result, many researchers have attempted to convert methane to syngas through the catalytic partial oxidation of methane (eqn (2)).8
| |
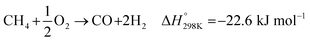 | (2) |
Although this conversion is only mildly exothermic, a small decrease in the selectivity of CO due to the total combustion of methane, which is a highly exothermic reaction, has resulted in a large increase in the reaction temperature. Furthermore, a high methane conversion together with a high space velocity can produce a large amount of heat in a small region of the catalyst. As it is difficult to remove this heat from the reactor, especially in equipment for large-scale purposes, the process can become difficult to control.9
Several researchers have proposed to reform methane catalytically with CO2 instead of steam, to obtain high selectivity of CO and more appropriate H2/CO ratios. They conducted the dry reforming of methane (DRM) (eqn (3)).10
| | |
CH4 + CO2 → 2CO + 2H2, ΔH°298 K = 247.0 kJ mol−1
| (3) |
This reaction bears important environmental implications for both the greenhouse gases, methane and carbon dioxide, which can be converted into valuable feedstocks.
The main setback of the DRM reaction is coke formation, which is caused by methane decomposition (eqn (4)) and the Boudouard reaction (eqn (5)). However, it has been reported that nickel-based catalysts can be suppressed by adding promoters in the deactivation process. In fact, strong Lewis bases (e.g., MgO, CaO) that are enhanced with the chemisorb of CO2, have resulted in the reduction of coke deposition in the reaction with C to form CO. Similar effects have been reported for lanthanide elements, such as CeO2 and La2O3, that can store and release oxygen, leading to the removal of carbon in the reaction between the deposited carbon and the lattice oxygen formed in these redox oxides.11
| | |
CH4 → C + 2H2, ΔH°298 K = 75.0 kJ mol−1
| (4) |
| | |
2CO → C + CO2, ΔH°298K = −172.0 kJ mol−1
| (5) |
The metal, nickel, is more commonly used as the active metal in the reforming process as its relatively abundant and its cost is low. However, the only drawback is that nickel easily induces the formation of carbon leading to catalytic deactivation.12 As a result, numerous research studies have been carried out to improve the catalytic activity and stability of nickel-based catalysts in the reforming process.13 The deactivation of nickel-based catalysts can be suppressed by the addition of promoters, such as strong Lewis bases (e.g., MgO, CaO) enhanced with the chemisorb of CO2, in order to reduce coke deposition in the reaction with C to form CO.14
It is noted that noble metals, such as Pt, Rh and Ru, are highly active towards DRM. Also, they are more resistant to the formation of carbon in comparison to other transition metals,15 Also, promoting Ni catalysts with noble metals that include Rh, Pt, Pd or Ru adds to the activity of the catalysts that show more stability against coke deposition when compared to the other non-promoted Ni catalysts.16 For example, bimetallic Ni–Pt that supports ZrO2 shows higher and more stable activity for a prolonged period of time than mono-metallic Ni/ZrO2. Hence, Ni–Pt shows potential in the industrial application for DRM.17 Meanwhilst, the activity and stability of bimetallic Ni–Pd catalysts are much higher than the mono-metallic Ni catalyst.18 These findings are consistent with the hypothesis that Pd helps in the prevention of the oxidation of Ni. A recent study over the promotion of Ni/MgO with Sn, Ce, Mn and Co reported higher catalytic activity and stability for Co-promoted catalysts; moreover, the coke deposition resistance was high for Sn and Co promoted catalysts. The higher catalytic activity for Co-promoted catalysts can be attributed to its high affinity for oxygen species enhancing it coke resistance properties. While Ce and Mn promoted catalysts exhibited lower catalytic performance, for Mn-promoted catalyst the agglomeration was the sole reason for its low catalytic activity and for Ce promoted catalyst lower activity was observed due to segregation of Ce as CeO2 due to its immiscibility with MgO which lead to the larger Ni0 particles.19
As such, the objective of this study has been to prepare a catalyst with high activity, selectivity, stability as well as the ability to prevent carbon deposition on the catalyst during the dry reforming of the methane reaction. The Pt, Pd, Ni/Mg1−xCexO catalysts were prepared by using the co-precipitation method that uses K2CO3 as a precipitant, followed by the impregnation of 1% Pt, 1% Pd, and 1% Ni using Pt(acac)2, Pd(acac)2, and Ni(acac)2, respectively. This was followed by a study into the comparison between the catalytic stability and coke formation. In addition, the study was also investigated the effects of the concentrations of CO2 and CH4, concentration of the catalysts, and the temperature of the conversion of the catalytic performance of the prepared catalysts in the dry reforming process as well as the study evaluated the stability of the catalyst. Finally, the study was examined the enhancement of the methane conversion of a stream of 1.25% oxygen gas passing through the reaction.
2. Experimental
2.1 Support and catalyst preparations
The catalysts, Mg1−xCexO (x = 0.00, 0.03, 0.07, 0.15), were prepared using the co-precipitation method as reported previously.20 Meanwhilst, the support MgO and promoter ceria Ce2O3 were prepared using a 0.1 M aqueous solution of Ce(NO3)3·6H2O (Merck; >99.0%) and Mg(NO3)2·6H2O (Merck; >99.0%) as the amount in the Table 1, and 1 M K2CO3 (Merck; >99.7%), which were used as the precipitants. The sample was washed with hot water after the filtration of the precipitant. Next, the sample was dried at 120 °C for 12 hours. Subsequently, it was pre-calcined in air at 500 °C for 5 h to remove CO2 from the precipitant. After that, the sample was pressed into disks at 600 kg m−2, and then calcined at 1150 °C for 20 h for enhancement of the mechanical properties and for ensuring good interaction between the support MgO and the promoter Ce2O3.
Table 1 Preparation of catalyst
| Catalysts |
Support (MgO) Mg(NO3)2·6H2O (g) |
Promoter (Ce2O3) Ce(NO3)3·6H2O (g) |
Total weight of MgO and Ce2O3 after calcine (g) |
Impregnation of the main catalyst (1% Pt) (1% Pd) (1% Ni) (g) |
| Pt(acac)2 |
Pd(acac)2 |
Ni(acac)2 |
| Pt/MgO |
25.0 |
0.0 |
1 |
0.02 |
0.029 |
0.044 |
| Pt, Pd, Ni/Mg0.97Ce0.033+O |
24.9 |
1.3 |
1 |
0.02 |
0.029 |
0.044 |
| Pt, Pd, Ni/Mg0.93Ce0.073+O |
23.8 |
3.0 |
1 |
0.02 |
0.029 |
0.044 |
| Pt, Pd, Ni/Mg0.85Ce0.153+O |
21.8 |
6.5 |
1 |
0.02 |
0.029 |
0.044 |
Table 1 shows the preparation of the catalyst. First, 1% Pt was impregnated using Pt(C5H7O2)2. H2O (Acros Chemicals; >99%) which was dissolved with dichloromethane for 5 h to produce Pt(acac)2/Mg1−xCexO. Finally, the catalysts, Pt, Pd, Ni(acac)2/Mg1−xCexO, were prepared by impregnating Pt(acac)2/Mg1−xCexO with a 1% of each Pd and Ni by using a solution of Pd(C5H7O2)2 (Aldrich; >99.5%) and Ni(C5H7O2)2·H2O (Acros Chemicals; >99%) in dichloromethane for 5 h, respectively. After impregnation in the air, the catalysts were dried for 12 h at a temperature of 120 °C. The dried catalysts were crushed and sieved to particles with 80–150 or 150–250 μm diameter.
2.2 Catalyst characterisation
The thermogravimetric analysis (TGA) was carried out on a Mettler Toledo TG-DTA Apparatus (Pt crucibles, Pt/Pt–Rh thermocouple) with the purge gas (nitrogen) flow rate of 30 ml min−1 and the heating rate of 10 °C min−1 from 50 to 1000 °C.
An X-ray diffraction analysis was performed using a Shimadzu diffractometer model XRD 6000. The diffractometer employed Cu-Kα radiation to generate diffraction patterns from powder crystalline samples at an ambient temperature. The Cu-Kα radiation was generated by Philips glass diffraction X-ray tube broad focus 2.7 kW type. The crystallite size D of the samples was calculated using the Debye–Scherrer's relationship.21 Where D was the crystalline size, λ was the incident X-ray wavelength, β was the full width at half-maximum (FWHM), and θ was the diffraction angle.
The Fourier transform infrared (FT-IR) analysis was carried out with the PerkinElmer spectrometer model 100 series (sample preparation UATR).
The total catalyst surface area was obtained using the Brunauer–Emmett–Teller (BET) method with nitrogen adsorption at −196 °C. The analysis was conducted using a Thermo Fisher Scientific S.P.A (model: Surfer Analyser) nitrogen adsorption–desorption analyser.
Transmission electron microscopy (TEM) (Hitachi H7100 TEM with accelerating voltage of 10 MV) was used to determine the crystal shape and the homogeneity of the catalysts.
Field Emission Scanning Electron Microscopy (FE-SEM) was used. The sample morphology was studied with the JEOL Field Emission scanning Electron Microscope (FE-SEM) model JSM 7600F at a very high magnification by using the field emission current. The particles of the samples were glued onto an aluminium stud by using double-sided tape. Then, it was coated with gold to make sure of the better visibility of the surface and to prevent electrical charging of the sample during analysis.
The active site of the catalysts was evaluated by Temperature Programmed Reduction (H2-TPR) using hydrogen conducted by Thermo Finnegan TPDRO 1100 apparatus Equipped with a thermal conductivity detector. In the reactor, about 0.05 g of the catalyst was placed and treated under 150 °C for 30 min in N2 (20 ml min−1). The analysis of hydrogen 5.51% in argon was carried out between 50 °C and 950 °C under argon flow (10 °C min−1, 25 ml min−1) and detected by a thermal conductivity detector.
XPS spectra were obtained using the Kratos Axis Ultra DLD system, equipped with a monochromatic Al Kα (1486.6 eV), dual X-ray sources (Al & Mg), an argon etching system for sample cleaning and depth profiling, parallel imaging XPS, AES, ISS and Vision software for controlling the system. The base pressure of the analyser chamber was 1 × 10−10 Torr. The excitation source, X-ray gun, was operated as a combination of 20 mA of emission current and 15 kV voltages. The hemispherical analyser was operated in the fixed analyser transmission (FAT) mode for both wide and narrow scanning. This value was set at 100 eV and 40 eV of the pass energy, respectively. The region of interest for the narrow scan corresponded to Mg2p, Ce3d, Pd3d, Ni2d, Pt4f and O1s photoelectron signals. The carbon charging correction refers to the binding energy of adventitious carbon at the binding energy of 285 eV. This highly sophisticated equipment is considered as a non-destructive analysis technique due to soft X-ray production to induce photoelectron emission from the sample surface. Therefore, the equipment was able to provide information about the surface layers or thin film structures (about the top 10–100 Å of the sample).
2.3 Catalytic evaluations
The catalytic evaluation for dry reforming of methane with CO2 (DRM) towards syngas (H2/CO) production as the model biogas reforming was carried out using a fixed bed stainless steel micro-reactor (i.d. Ø = 6 mm, h = 34 cm). The reactor was connected to a mass flow gas controller (SIERRA instrument) and an online gas chromatography (GC) (Agilent 6890N; G 1540N) equipped with Varian capillary columns HP-PLOT/Q and HP-MOLSIV. Prior to the reaction, approximately 0.02 g of the catalyst was reduced by flowing 5% H2/Ar (30 ml min−1) at 700 °C and holding for 3 h. The reforming reaction was performed by flowing the feed, a gas mixture consisting of CH4/CO2 in (2/1) and (1/1) mol, at a rate of 30 ml min−1. The reforming has been studied from 700 to 900 °C at 1 atm, then holding was carried out for 10 h (1 atm, GHSV = 30 ml min−1).
The reduction step was aimed to reduce the (Ni2+, Pd2+, Pt2+) phase of the catalyst to the metals (Ni0, Pd0, Pt0) phase at the active site of the catalysts. The tested catalyst was placed in the middle of a reactor vertically and held in place by plugs of quartz wool. In order to control and ensure the reaction temperature, a thermocouple was placed into the catalyst chamber. The calculations for the CH4 and CO2 conversions, H2 and CO selectivity, as well as syngas (H2/CO) ration were defined as the following equations (eqn (6)–(10)).
| |
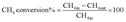 | (6) |
| |
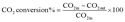 | (7) |
| |
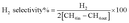 | (8) |
| |
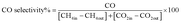 | (9) |
| |
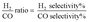 | (10) |
3. Results and discussion
3.1. Characterisation of the catalysts
3.1.1 XRD patterns. Fig. 1a–d illustrates the XRD patterns of the catalysts with magnesium and cerium contents. The diffraction peaks were recorded at 2θ = 37.0 (111), 42.9 (200), 62.3 (220), 74.7 (311) and 79.1° (222). This was due to the cubic form of magnesia (JCPDS file no: 00-002-1207). Meanwhilst, the diffraction peaks recorded at 2θ = 28.6 (111), 33.1 (200), 47.5 (220), 56.4 (311), 59.1 (222), 69.4 (400), 76.7 (331) and 79.1° (420) were due to the cubic form of ceria (JCPDS file no.: 00-034-0394). The peaks were recorded at 2θ = 47.7 (116), 56.5 (115), 59.3 (304), 62.5 (104), 69.7 (224), 77.0 (317), and 79.3° (318) were attributed to the cubic form of catalyst complex (Ce–Mg–O). However, there were no diffraction peaks for the catalyst of 1% platinum, palladium, and nickel in all the patterns. This was because the amount of these elements was very small. This observation agrees with the results reported by Grange.22 The average crystalline size was identified through the diffraction of the highest peak in the XRD patterns that used the Debye–Scherrer equation (Table 2). The findings show that the size of the crystal was inversely proportional to the increasing amount of ceria in the catalysts. This phenomenon could be linked to the effects of platinum, palladium, and nickel that remained on the surface of the sample that inhibited the growth of magnesia crystallites. The size of the crystal was recorded at 42.2, 53.3, 48.7, and 48.0 nm for Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O, respectively. Clearly, the predominant crystal system for all the samples was a cubic one. This is supported by TEM and FESEM, which showed cubic shaped particles, as well. In another study, Abimanyu et al.23 recorded the XRD pattern of MgO–Ce2O3 catalyst and found 38.0 nm.
 |
| | Fig. 1 XRD patterns of the catalysts (a) Pt, Pd, Ni/MgO (b) Pt, Pd, Ni/Mg0.97Ce0.033+O (c) Pt, Pd, Ni/Mg0.93Ce0.073+O (d) Pt, Pd, Ni/Mg0.85Ce0.153+O. | |
Table 2 Particles size measurement by XRD, TEM and XRF results
| Catalysts |
TEM (nm) |
Crystal size (D) Debye Sherrer eq. (nm) |
Ni% |
Pd% |
Pt% |
Mg & Ce% |
| Pt, Pd, Ni/MgO |
56 |
42.2 |
1.13 |
1.09 |
1.15 |
96.30 |
| Pt, Pd, Ni/Mg0.97Ce0.033+O |
68 |
53.3 |
1.18 |
1.11 |
1.19 |
96.17 |
| Pt, Pd, Ni/Mg0.93Ce0.073+O |
59 |
48.7 |
1.07 |
1.10 |
1.15 |
96.2 |
| Pt, Pd, Ni/Mg0.85Ce0.153+O |
61 |
48.0 |
1.13 |
1.22 |
1.26 |
96.1 |
For the elemental analysis of all the components in the catalyst, XRF has been used. Table 2 shows that the Ni, Pd, and Pt percentages were slightly more than 1, which was due to the incomplete precipitation of the magnesium and cerium metal precursors in the method of co-precipitation. This had a slight effect on the results.24
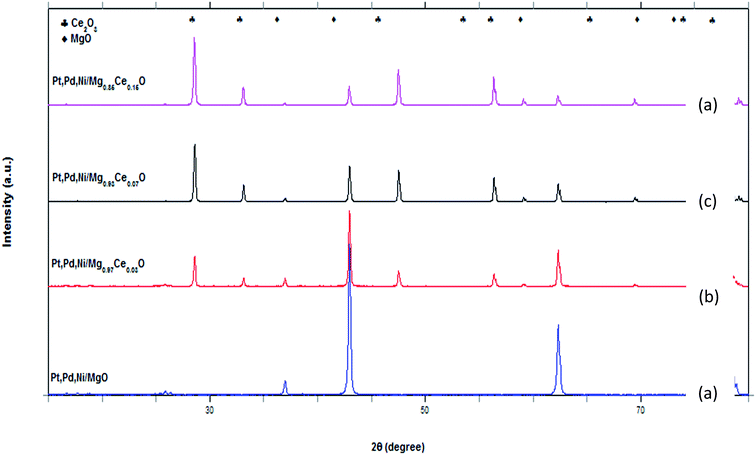
3.1.2 XPS analysis. Fig. 2a–e shows the X-ray photoelectron spectroscopy (XPS) that has been used to investigate the elements O1s, Mg2p, Ce3d, Ni2p, Pd3d and Pt4f of the reduced catalyst Pt, Pd, Ni/Mg0.93Ce0.07O. In the study of the surface of a few layers of the catalyst, measuring 3–12 nm, the spectra of the XPS revealed a few findings. In Fig. 2a, there are five distinct oxygen species for O1s. They are on the surface of the catalyst assigned to Ni–O, Ce–O, Mg–O, Pt–O and Pd–O at a binding energy of 528.5, 529.5, 532, 533 and 534 eV, respectively. In Fig. 2b, there are two distinct peaks, obtained from Mg2p, bulk Mg–O and MgCO3 at a binding energy of 49 and 51 eV, respectively. Meanwhilst, Fig. 3c shows the Ce3d of Ce2O3. It took the range of binding energy from 925–875 eV. The highest photoelectron signal intensity in the high binding energy region is shown in Ce–O. The peaks have been recorded at 887.5 eV and 882.5 eV, respectively. The Ni2p3/2 spectrum measures 30 eV wide. It consists of four main peaks obtained from the presence of Ni–O (Fig. 2d). Pd3d and Pt4f give four peaks for each due to Pt4f5/2 and Pt4f7/2 appearing at around 75–65 eV in the catalyst (Fig. 2e–f).25,26
 |
| | Fig. 2 XPS narrow scans of the reduced catalyst. (a) O1s (b) Mg2p (c) Ce3d (d) Ni2p (e) Pd3d (f) Pt4f. | |
 |
| | Fig. 3 TPR-H2 profiles of catalysts reduced in a (5% H2/Ar) stream at a temperature ramp of 10 °C min−1. | |
The analysis of XPS indicates that the solid solution of Ce2O3–MgO shows a low and a high binding energy of 51 eV and 882.5 eV for Mg2p and Ce3d, respectively. As a result, it is imperative to transfer the electrons from Ce2O3 to MgO. This is to slow down the reduction of Ce2O3 during the preparation of the reduced catalyst. However, during the slowdown, there is an increase in the interaction between the two oxides. This leads to the segregation of Ce atoms as small particles on the surface of the catalyst, resulting in a high dispersion of Ce, which is responsible for the high level of activity of the catalyst. Furthermore, the segregated Ce particles, which have been extracted from the substrate, interact strongly with the Ce remaining on the substrate, resulting in the attenuation of their sintering. In this case, there is no formation of coke due to the high dispersion of Ce on the surface of the catalyst. Also, the Ce clusters are not big enough for the formation of coke. The TEM has indicated that a highly effective Ce2O3–MgO solid solution contains crystallite sizes of about 80 nm.18
3.1.3 H2-TPR. The TPR experiments were conducted on the following catalysts: Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O to investigate their reduction behaviour. Fig. 3a–d and Table 3 show the TPR profiles of these catalysts. Fig. 4a shows three well-defined reduction peaks in the TPR profile of Pt, Pd, Ni/MgO. The first reduction peak has been recorded at 130 °C. This is attributed to the reduction of the PtO species in the production of Pt0, as compared to the previous study of Mahoney et al.27 in which it was detected at 114 °C. The second reduction peak is centered at 184 °C. This is due to the reduction of PdO to Pd0. Finally, the third peak was in the region of 511 °C. This is associated with a reduction in the NiO species that led to a strong interaction with the supporting material to produce Ni; whereas, Bao et al.28 found the reduction temperature of NiO in the Ni/CeMgAl catalyst at 516 °C.
Table 3 TPR-H2 values of the different catalysts
| Catalysts |
Temp. °C |
Temp. °C |
Temp. °C |
Temp. °C |
Temp. °C |
Amount H2 gas adsorbed (μmol g−1) |
| Pt, Pd, Ni/MgO |
130 |
184 |
511 |
— |
— |
488.6 |
| Pt, Pd, Ni/Mg0.97Ce0.033+O |
129 |
169 |
481 |
502 |
711 |
429.1 |
| Pt, Pd, Ni/Mg0.93Ce0.073+O |
132 |
198 |
495 |
508 |
727 |
941.9 |
| Pt, Pd, Ni/Mg0.85Ce0.153+O |
135 |
160 |
490 |
510 |
735 |
737.7 |
 |
| | Fig. 4 TEM image of catalysts (a) unreduced Pt, Pd, Ni/MgO (b) unreduced Pt, Pd, Ni/Mg0.97Ce0.033+O (c) unreduced Pt, Pd, Ni/Mg0.93Ce0.073+O (d) unreduced Pt, Pd, Ni/Mg0.85Ce0.153+O (e) reduced Pt, Pd, Ni/Mg0.93Ce0.073+O with 5% H2 in Ar at 700 °C (f) Pt, Pd, Ni/Mg0.93Ce0.073+O after 200 h reaction, at 900 °C, and CH4/CO2 ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1. | |
Fig. 3b–d and Table 3 illustrate the TPR profile for the catalysts including the promoter Ce2O3. The TPR profiles for Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O catalysts are quite different from the catalyst Pt, Pd, Ni/MgO. The findings show five peaks. The first three peaks of the catalyst Pt, Pd, Ni/Mg0.97Ce0.033+O were recorded at 129 °C, 169 °C, and 481 °C; whilst the peaks of the catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O were recorded at 132 °C, 198 °C, and 495 °C. Meanwhilst, the peaks of the catalyst Pt, Pd, Ni/Mg0.85Ce0.15O were recorded at 135 °C, 160 °C, and 490 °C. This was due to the reduction of PtO, PdO, and NiO on the surface of the catalysts to obtain the elements Pt0, Pd0, and Ni0, respectively. The fourth peak of the catalysts Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O was found where the temperatures were recorded at 502 °C, 508 °C, and 510 °C, respectively. The findings correspond to the reduction of Ce2O3 on the surface. In fact, there was a significant lowering of the surface of Ce2O3 when there was a reduction in the temperature of the catalysts. There are several possible explanations for this phenomenon. Firstly, it could be due to the improved dispersion of Ce2O3 particles during the incorporation of MgO into Ce2O3 and the retardation of sintering.29 Secondly, it could be due to the strong interaction between Ce2O3 and Pt, Pd, and Ni metals; these occur during the overlapping of the PtO, PdO and NiO and Ce2O3 in the reduction of peaks. The fifth peak was recorded at temperatures of 711 °C, 727 °C, and 735 °C. This is attributed to the reduction of bulk Ce2O3 that led to strong interactions between the species of the promoter, Ce2O3, and the support, MgO. It has been revealed that the catalysts show more reducibility with an increase in the loading of the promoter. This finding is in agreement with the results of previous research studies, as compared with the previous study of the cerium reduction results of Rotaru et al.,29 where the reduction of cerium took place at 490 °C and 790 °C. There is a good dispersion of promoters to the support, and it induces a high level of interaction between the support with doping Pt, Pd, and Ni species. Indeed, the significant peak of the TPR profile recorded at temperatures between 684–737 °C proves that Ce2O3 alone can reduce the range in temperatures.30 It is also obvious that the addition of the promoter, Ce2O3, is effective in the reducibility of catalysts with MgO support. This could be attributed to the acidic–basic properties of the support. It has been found that Mg1−xCex3+O with a higher basicity than MgO interacts more with the Ce2O3 promoter. Thus, the reductions in PtO, PdO, and NiO are more obvious. This is due to the redox property of Mg1−xCex3+O.31
The total amount of H2-consumption in the reduction of Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O was calculated from the total area of the peaks. The calculations for the catalysts were 488.6, 429.1, 941.9, and 737.7 μmol g−1 catalyst, respectively. Based on the TPR-H2 results, the catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O shows that it is the most active site amongst the other catalysts. In other words, it is the best catalyst for dry reforming of methane.
3.1.4. BET. Table 4 lists the surface area of BET, pore volume and pore radius of the support MgO for the different catalysts: Pt, Pd, Ni/Mg1−xCexO (where x = 0.00, 0.03, 0.07, and 0.15). The surface area of BET for the catalysts Pt, Pd, Ni/MgO with a cubic structure supported with TEM was 12.4 m2 g−1 whilst the surface area of BET for the support MgO was 11.1 m2 g−1. The higher reading for the former was due to the effects of Pt, Pd, and Ni loading on the specific surface area of the support MgO. In this case, the BET surface areas of the catalysts Pt, Pd, Ni/MgO was considerably lower than the BET surface areas of the conventional catalysts: Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O and Pt, Pd, Ni/Mg0.85Ce0.153+O which were recorded at 12.7, 19.8 and 12.9 m2 g−1, respectively. This is because the magnesia pores were partially covered by the layer of Pt, Pd, and Ni particles. However, the BET surface area of the MgO, that was promoted by Ce2O3 was almost similar to the one of the conventional catalysts Pt, Pd, and Ni with binary support.32 In addition, the characteristics of the supported Pt, Pd, and Ni catalysts with a cubic structure included extremely low metal dispersion and small surface areas of the Pt, Pd, and a few Ni particles. This might be due to the strong interaction between the layers of Pt, Pd, and Ni and the support of MgO with the promoter Ce2O3. The pore volume of the catalyst, Pt, Pd, Ni/Mg0.85Ce0.153+O, was recorded at 0.22 cm3 g−1. This value was slightly bigger than the value of the other catalysts which was recorded at 0.21 cm3 g−1. This is in contrast to the study of Bao et al. (2015) where the pore volume of the NiCeMgAl catalyst was found to be 0.51 cm3 g−1.28
Table 4 The main textural properties of fresh catalysts
| Sample name |
Specific surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore radius (oA) |
| MgO |
11.1 |
0.21 |
9.9 |
| Pt, Pd, Ni/MgO |
12.4 |
0.21 |
9.7 |
| Pt, Pd, Ni/Mg0.97Ce0.033+O |
12.7 |
0.21 |
13.2 |
| Pt, Pd, Ni/Mg0.93Ce0.073+O |
19.8 |
0.22 |
12.5 |
| Pt, Pd, Ni/Mg0.85Ce0.153+O |
12.9 |
0.21 |
12.4 |
| Spent catalyst |
20.2 |
0.23 |
15.6 |
Table 4 shows the pore radius of the different catalysts. The pore size of the support MgO was recorded at 9.9 Å; whereas, the pore size of the catalyst Pt, Pd, Ni/MgO was recorded at 9.7 Å. The pore radius of the remaining catalysts was inversely proportionate to the increase in the support of the promoter, Ce2O3; whereby, the pore radii of the catalysts: Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O and Pt, Pd, Ni/Mg0.85Ce0.153+O were recorded at 13.2 Å, 12.5 Å, and 12.4 Å, respectively.33 This shows that the catalyst Pt, Pd, Ni/Mg0.93Ce0.07O with a high surface area was able to perform better in the dry reforming of the methane reaction as compared to the other catalysts.
3.1.5. TEM. Fig. 4a–d shows the TEM images of the catalysts Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O with cubic structures. The catalysts were calcined at 1150 °C with a uniform particle distribution in the absence of free Ce2O3. Fig. 4b–d can be confirm the formation of the MgO–Ce2O3 solid solutions34 with cubic oxide particles on the Pt, Pd, and Ni layers of the supported metal. The catalyst Pt, Pd, Ni/Mg0.97Ce0.033+O (Fig. 4b) was well dispersed with 1% of the Pt, Pd and Ni metal particles, each for the support magnesia-ceria of sizes ranging from 45 to 85 nm.35 In addition, the TEM analysis of the catalyst, Pt, Pd, Ni/Mg0.85Ce0.153+O, indicated that an agglomeration of the nanoparticles at a specific distance between the metal crystallites had induced growth. Generally, metallic platinum, palladium and nickel, are known to catalyse this type of growth.36 Although the size distribution obtained from the images of TEM were more realistic and accurate. The TEM results (Fig. 4a–e) are well in relation with XRD data which showed that the complex Mg–Ce–O were also cubic in nature like MgO and Ce2O3.
3.1.6. Thermal analysis study. Fig. 5a–d shows the analysis of the TGA for the reduced catalysts, Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O. The findings indicate a weight loss; but, at only one stage of the thermal process. The weight loss was recorded at about 2% at temperatures ranging from 100 °C to 120 °C. This could be attributed to the removal of moisture from the catalysts Pt, Pd, Ni/Mg1−xCexO (Fig. 5a–d). Meanwhilst, the weight loss was recorded at 1.5%, 2.2%, 1.8%, and 1.6% for the catalysts: Pt, Pd, Ni/MgO, Pt, Pd, Ni/Mg0.97Ce0.033+O, Pt, Pd, Ni/Mg0.93Ce0.073+O, and Pt, Pd, Ni/Mg0.85Ce0.153+O, respectively. On the other hand, the Pt, Pd, Ni/Mg0.85Ce0.153+O and Pt, Pd, Ni/Mg0.93Ce0.073+O, catalysts (Fig. 5c–d), showed an additional second stage. Here, the weight loss was recorded at 2% and 3%, respectively, which may be attributed to the loss of oxygen atoms from the catalysts. The graph shows that initially, the entire weight of the compound increased slightly. This was due to the adsorbing of the compound to the N2 gas in the machine. All the compounds became thermally stable at a temperature of 500 °C. This was because of the high melting point of magnesia and ceria at 2852 °C and 2177 °C, respectively. Fig. 5a–d reveals that the components of the catalyst show good interaction components. This is in agreement with the results of Mojovic et al.37
 |
| | Fig. 5 TG of the catalysts (a) Pt, Pd, Ni/Mg0.97Ce0.033+O (b) Pt, Pd, Ni/MgO (c) Pt, Pd, Ni/Mg0.85Ce0.153+O (d) Pt, Pd, Ni/Mg0.93 Ce0.073+O. | |
3.2. Catalytic performance in biogas reforming
3.2.1. Effect of the reactant concentration on conversion. The reaction activity of the dry reforming of methane was indicated by the conversion of CH4 and CO2, and the selectivity was expressed in terms of the H2/CO ratio. When the temperature was set above 900 °C, the findings of the blank tests (reaction without catalyst) revealed the presence of H2 and CO in the outlet gas. This may be due to the decomposition reaction of methane (eqn (5)). When Mg1−xCexO was used without Pt, Pd, and Ni metals, the conversion of CH4 and CO2 was very low, recording 38% and 48%, respectively; whilst, the H2/CO ratio was recorded at 0.2%. These results indicate that the reaction on the pores of the support promoter was weak. Likewise, the BET results also showed that there were pores in the catalyst. On the other hand, when the catalysts Pt, Pd, Ni/Mg1−xCex3+O were used, the conversion of CH4 and CO2 and the ratio of H2/CO showed an increase (Fig. 6). This means that the Pt, Pd, and Ni metals doped on the support play a crucial role in the catalytic reaction. The catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O recorded a CH4 and CO2 conversion in 83% and 97% for CH4:CO2 (2:1), respectively, and the H2:CO ratio of 1.15. However, the conversion of the gases, CH4:CO2 at the ratio of (1:1) was 84%, 99% and 1.2, respectively. This indicates that the ratio of 1:1 provides the best resistance to the deactivation of the catalyst. The reason for this is the carbon formation and high selectivity of H2 and CO (Fig. 6). The other catalysts have also been shown to be similar in this aspect.38
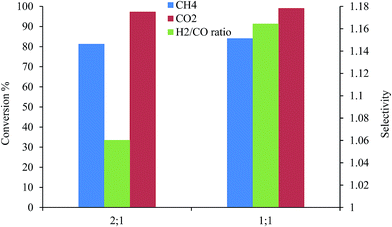 |
| | Fig. 6 The effect of changing the ratio concentration of CH4:CO2 reactant 1–2:1 and 2–1:1 over the % of their conversion and H2/CO ratio for Pt, Pd, Ni/Mg0.93Ce0.073+O catalyst at 900 °C. | |
3.2.2. Effect of the catalyst concentration on conversion. Fig. 7 and Table 5 show the effects of the concentration of the catalyst during the conversion process. The conversion of CH4, CO2 and the ratio of H2/CO were in an ascending order as in: Pt, Pd, Ni/MgO < Pt, Pd, Ni/Mg0.97Ce0.033+O < Pt, Pd, Ni/Mg0.85Ce0.153+O < Pt, Pd, Ni/Mg0.93Ce0.073+O.
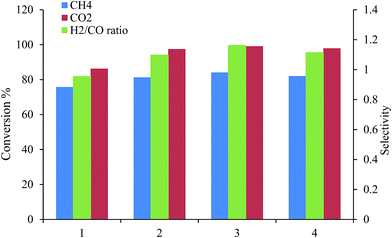 |
| | Fig. 7 The effect of using different catalysts (1) Pt, Pd, Ni/MgO, (2) Pt, Pd, Ni/Mg0.97Ce0.033+O, (3) Pt, Pd, Ni/Mg0.93Ce0.073+O, and (4) Pt, Pd, Ni/Mg0.85Ce0.153+O on CH4, CO2 conversion and H2/CO ratio at 900 °C for the 1:1 ratio of CH4:CO2. | |
Table 5 XRF analysis for the catalysts
| Sample name |
Ni% |
Pd% |
Pt% |
Mg & Ce% |
| Pt, Pd, Ni/MgO |
1.23 |
1.09 |
1.15 |
96.30 |
| Pt, Pd, Ni/Mg0.97Ce0.033+O |
1.18 |
1.11 |
1.19 |
96.17 |
| Pt, Pd, Ni/Mg0.93Ce0.073+O |
1.07 |
1.10 |
1.15 |
96.7 |
| Pt, Pd, Ni/Mg0.85Ce0.153+O |
1.13 |
1.22 |
1.26 |
96.1 |
The Pt and Pd were combined with nickel on the support, MgO–Ce2O3. The experiments were conducted under the following conditions: a temperature of 900 °C and 1 atm with a feed ratio (CH4:CO2) of 1:1 (Fig. 7 & Table 5). As for the conversion of methane, the highest conversion recorded was found in the catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O (84%); whilst, the lowest conversion was observed in the catalyst Pt, Pd, Ni/MgO (75%). Another finding is that most of the catalysts tested showed a slight deactivation after 200 h. Overall, the conversion of CO2 was more stable than for methane; and, for the conversion of CH4, the highest conversion was found in the catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O (99%); whilst, the lowest conversion was observed in the catalyst Pt, Pd, Ni/MgO (86%). From the readings, it can be concluded that the best catalyst is Pt, Pd, Ni/Mg0.93Ce0.073+O.
The product ratio of H2/CO over all the tri-metallic magnesia-ceria catalysts (Fig. 7 & Table 5) was recorded at above 1. This indicated that the process of CO2 conversion of the Ni metal was less favourable than that of the tri-metallic catalysts as compared to the other studies.28 Also, the side reactions showed improvement. This is evident from the observed difference between the conversions and the product yields. Table 5 shows that with an increase in the concentration of ceria, there were increases in the conversion rate of CH4 and CO2 as well as the ratio of H2/CO. The best result observed by the catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O was found at the most active site of H2-TPR and from the large surface area indicated from the BET result.
This phenomenon reveals that the incorporation of Ce2O3 into the MgO catalysts can significantly depress the Reverse Water Gas Shift (RWGS) reaction (eqn (11)).
| | |
CO2 + H2 → CO + H2O, ΔH°298K = 41.0 kJ mol−1
| (11) |
The results also indicate that the rate of the CO formation in the dry reforming of methane was dependent on the strong interaction between the promoter, Ce2O3, and the support, MgO solid solution, based on the following findings: the ratio of 0.07:0.93 mole in the catalyst, the largest surface area of 19.8 m2 g−1 (Table 4), and the most active site of 941.9 μmol g−1 from the total amount of H2-consumption in the H2-TPR study (Table 3). Hence, the formation of a solid solution was crucial in the generation of the active sites for the CO2 reforming of methane.
This took place because the entire promoter, Ce2O3, was present as a solid solution, which stabilised both oxides. Only the surface layer of Ce2O3 of the solid solution of the catalyst Ce2O3–MgO was reduced during the reduction of hydrogen at 700 °C. Furthermore, the Ce sites generated remained in close contact with the solid solution, which became a hindrance to the Ce sintering.39 Furthermore, the sites that were responsible for the catalytic process can be found in the Pt, Pd, and Ni particles. This is because the Pt, Pd, and Ni particles have a strong interaction with MgO–Ce2O3. When there was an increase in the concentration of Pt, Pd, and Ni in the support, there were no significant changes in the CH4 and CO2 conversion and selectivity. This can be attributed to the formation of nanoparticles as a result of the XRD results (see Debye Sherrer's equation) and TEM results (see Table 2). It is known that the size distribution obtained from TEM images tends to be more realistic and accurate. However, the images have presented some limitations. On the one hand, X-ray diffraction provided a very simple possible estimation of the crystal size from the broadening of the XRD reflections through the use of Scherrer's formula. On the other hand, nanoparticulation of the particles is a preferred choice. The use of the selected nanoparticles as catalysts for this research study not only maximised the surface area but secured more reaction. Also, it ensured a good dispersion of the Pt, Pd, and Ni metals on the surface of the catalysts, as well as strong Lewis basicity with metal oxide support. The increase of the support in Lewis basicity enhanced the ability of the catalyst to chemisorb CO2 in the dry reforming of methane. Also, the adsorbed CO2 reacted with C to form CO (eqn (12)), resulting in the reduction in coke formation.
| | |
CO2 + C → 2CO, ΔH298K° = +172 kJ mol−1
| (12) |
The formation of the Ce2O3–MgO solid solution served as a unique approach in the prevention of carbon deposition. MgO is a strong Lewis base, with a high adsorption of CO2 in reducing or preventing carbon deposition. Furthermore, the XPS results revealed that the reduction of Ce2O3 in the solid solution of Ce2O3–MgO was much more difficult, often leading to the formation of smaller particles of cerium on the surface than that of pure Ce2O3.40,41 The combination of the surface basicity and the particle size of the small metals made-up the ability of the catalyst, MgO-based solid solution, in the prevention of carbon deposition. Although the size distribution obtained from the TEM images tended to be more realistic and accurate, there were some setbacks in the images.
Furthermore, the conversion of CH4 and CO2 was very high due to the particle size involved in the reactive activity. The doping metals, Pt, Pd, and Ni, were prepared based on the Debye Sherrer equation, and supported by the TEM analysis. The metal size had to be as minute as nanoparticles. Thus, it is evident that particle size plays a significant role in the activity of the reaction. An increase in the conversion of the reactants and selectivity (yield) was mainly due to the reduction of the particles in nano-ranged sizes leading to an increase in active sites which was recorded at 941.9 μmol g−1, and in the surface area which was recorded at 19.8 m2 g−1 (refer to Tables 3 and 4).
3.2.3. Effect of temperature on conversion. Fig. 8 shows the result of the activity and selectivity of catalyst Pd,Pd,Ni/Mg0.93Ce0.073+O at temperatures ranging from 700 °C to 900 °C. Generally, the conversion of CH4:CO2 (1:1) increased as the temperature was raised from 700 °C to 900 °C. This was because the dry reforming of the methane reaction showed a strong endothermic reaction (eqn (3)) and a higher temperature increased the conversion rate. This has been observed in previous studies.42 When the temperature was increased from 700 °C to 900 °C, the CH4 conversion of Pd,Pd,Ni/Mg0.93Ce0.073+O showed an increase from 60% to 84%; whilst the CO2 conversion showed an increase from 38% to 99%. At temperatures above 900 °C, there was no significant increase in the conversions of CH4 and CO2. Fig. 8 shows the H2/CO ratio of the catalyst at various temperatures; when the temperature was <900 °C, the H2/CO ratio of the samples was recorded as <1. This was because the reverse water gas shift reaction (RWGS), (eqn (11)) consumed additional H2 and produced CO; thereby lowering the H2/CO ratio. At a temperature of 900 °C, the H2/CO ratio of Pd,Pd,Ni/Mg0.93Ce0.073+O was recorded at 1.16, indicating a smaller contribution from the RWGS reaction (eqn (11)).15
 |
| | Fig. 8 The influence of temperature on the catalytic activity of the Pt, Pd, Ni/Mg0.93Ce0.073+O catalyst. (1) 700 °C (2) 800 °C (3) 900 °C for the 1:1 ratio of CH4:CO2. | |
3.2.4. Stability tests. Fig. 9 shows the reading of the temperature tests. The findings revealed that when the temperature was at 900 °C, the conversion for both CH4 and CO2 was high. Generally, in this mechanism, a molecule of methane reacts on the surface of Ni to produce desorbed hydrogen and hydrocarbon species CHx (x = 0–4); if x = 0, the carbon deposition on the Ni metal surface43 is indicated, as in the mechanism shown below:
| CH4 + 2Ni(as) → CH3Ni(as) + HNi(as) |
| CH3Ni(as) + Ni(as) → CH2Ni(as) + HNi(as) |
| CH2Ni(as) + Ni(as) → CHNi(as) + HNi(as) |
| CHNi(as) + Ni(as) → CNi(as) + HNi(as) |
| 2HNi(as) → H2(g) + 2Ni(as) |
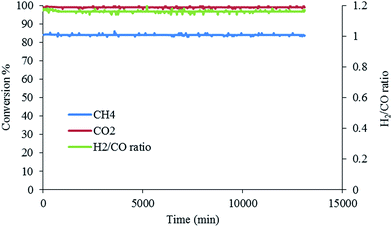 |
| | Fig. 9 Stability tests of Pt, Pd, Ni/Mg0.93Ce0.073+O catalysts at 900 °C for the 1:1 ratio of CH4:CO2, for 200 h. (GHSV = 15000 ml gcat−1 h−1, atmospheric pressure). | |
Nakamura et al.44 stated the effects of a promoter in the catalyst on the dry reforming of methane. One of them was by increasing the dispersion of Pt, Pt and Ni, carbon dioxide was activated on the support-promoter in proximity to the metal particle to form a carbonate species. Following that, the CHx species reduced the carbonate to form carbon monoxide (CO).
| CO2(support) + O2−(support) → CO32−(support) |
| CO32−(support) + 2H(support) → HCO2−(support) + OH−(support) |
The role of the promoter Ce2O3 in the catalyst was to provide a highly stable platform and a strong resistance to coking that takes place during the conversion of CO2 and CH4 as well as a H2/CO ratio for over 200 h of reaction. The formation of carbon on the catalyst during the dry reforming of the methane reaction was removed by Ce2O3. Also, the CO2 adsorption was enhanced in the presence of Ce2O3 as Ce2O3 can increase the basicity. The next stage after adsorption, was the formation of the carbonate species. It occurred mostly on Ce2O3; whereby, it dissociated to CO2 and then to CO and O. Then O atom is transferred to Ce promoter and finally combine with the C which was deposited on the metal catalyst to produce CO.45 It was evident that there was remarkable decrease of carbon deposition on the catalyst. When the concentration of the Ce2O3 was low, the CO2 conversion showed an increase in the formation of strong ionic oxides, Ce2O2CO3, which resulted in the attraction of CO2 to the surface of the catalyst thereby increasing CH4 conversion. When the concentration of Ce2O3 was high, the conversion of both CH4 and CO2 showed a decrease. This probably took place because of an increase in the electron density of Pt, Pd, and Ni.46 The Ce2O2CO3 species participated directly in DRM by decomposing to produce CO and to provide an oxygen species to react with the carbon deposits on the interface of Pt, Pd, and Ni–Ce2O2CO3; thus, restoring the activity of the Pt, Pd, and Ni sites. Similarly, the Ce2O3 supported catalysts can facilitate the dissociation of adsorbed CO2. In addition to its promotional effect on CO2, the dissociative adsorption of ceria can also improve the dispersion and stabilisation of small metal particles. In fact, ceria is one of the oxides known to exert strong interactions on the supported metallic phase, resulting in significant alterations of the surface properties of both the oxide and metal.47
| CO2(g) → CO(support) + O(promoter) |
| C(metal) + O(promoter) → CO2(g) |
The activity and stability of the tri-metallic catalyst Ni–Pd–Pt was much higher than that of the mono-metallic catalyst Ni, or bi-metallic catalysts Pd–Ni and Pt–Ni. This is consistent with the hypothesis that Pt and Pd help prevent oxidation of Ni due to an increase in its electron density.48 The formation of a Pt–Ni or Pd–Ni bi-metallic cluster increases the reducibility of Ni, resulting in higher and more stable activity for experiments of over 200 h.49
3.2.5. Post-reaction characterization. The TEM images and TGA analysis detected the presence of a coke deposit with an oxygen stream of spent catalyst. Fig. 4f shows the TEM images. The images show that the original structure of the catalyst was maintained even after 200 h of stream testing. Furthermore, the spent catalyst kept its two-dimensional cubic texture. However, there was an obvious increase in pore size from 12.2 Å to 15.6 Å, in the spent catalyst. The BET analysis also showed that the surface area of the spent catalyst was slightly increased from 19.8 to 20.2 m2 g−1. The phenomenon of a slight metal sintering was observed in the spent catalyst. Whilst the two-dimensional cubic channel of spent catalyst limited the sintering of the active metals inside the pore, the active metals supported on the outside surface experienced significant sintering. Since no filamentous carbon was found in the spent catalyst, it can be concluded that the coke deposition was negligible.Fig. 10 shows the TGA results with an oxygen stream of post-reaction tri-metallic catalyst Pt, Pd, Ni/Mg0.93Ce0.073+O together with the calculation of weight change at each temperature range illustrated at the thermo-gram. There are altogether three different regions of temperature. The first region is the low temperature range. Here, the spent catalyst experienced an increase in weight. The second region is the mid temperature range. The spent catalyst experienced a decrease in weight in this region. The third region is the high temperature range. The spent catalyst found here experienced an increase in weight. The oxidation of the Ni particles at temperatures above 100 °C caused the increase in weight. Zhu et al.50 reported the increase in weight of the spent catalyst as less than 1%. The compound was found to be stable in the range of the temperatures (150–500) °C; whilst the loss of weight at a temperature of 650 °C was due to the oxidation of deposited carbon. It was found that the carbon deposited on the spent catalyst was layered carbon, with further support in the absence of filamentous carbon from the TEM results of the spent catalysts. The amount of coke deposited on the spent catalyst was calculated to be 2.4 wt%. Finally, all elements on the surface were synthesized by oxygen after the removal of carbon on the surface as CO2. In conclusion, the results showed that at 900 °C, there was little deposition of coke on the surface of the catalyst, and the formation of the coke was related to the dispersion of metal in the catalysts. The size of the smaller crystal metal catalysts makes it less prone to deactivation.
 |
| | Fig. 10 TGA profiles of spent Pt, Pd, Ni/Mg0.93Ce0.073+O catalyst (20 mL min−1 O2 stream under a temperature ramp of 10 °C min−1). | |
3.2.6. Improvement in the stability and selectivity of the catalyst. The dry reforming of the methane reaction can be enhanced by conducting experiments in concentrations of low oxygen flow (1.25%). Fig. 11 shows enhancement in the conversion of CH4, from 84% to 96%, due to the addition of an oxidant (O2) to partially or completely synthesize the methane as well as the use of exothermicity of the reaction to supply the necessary heat directly to the DRM reactant mixture.46 However, the CO2 conversion and the H2/CO ratio are not affected. This may be due to the reaction of oxygen with CH4 to produce CO and H2O (eqn (13)). Finally, the steam reacts with the deposited carbon to produce syngas (eqn (14)). Furthermore, O2 can reduce coke deposition on the catalyst (eqn (15)). Therefore, this process has reduced the deposition of carbon; consequently improving the life time of the catalyst.| | |
CH4 + 1.5O2 → CO + 2H2O(v)
| (13) |
| | |
C(s) + H2O → CO + H2
| (14) |
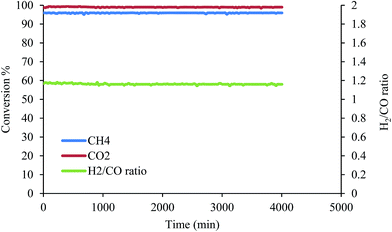 |
| | Fig. 11 DRM reaction of the Pt, Pd, Ni/Mg0.93Ce0.073+O catalyst under 900 °C with 1.25% O2. | |
4. Conclusions
It is inevitable that biogas is an attractive carbon source for the production of clean fuels and chemicals because it is renewable, easily available, and inexpensive. In addition, its processing, storage and usage employs technologies and infrastructure that are developed for natural gas. Meanwhilst, the dry reforming of biogas is a promising technology in the production of syngas. In terms of synthesized carbon, this approach is highly beneficial, as both major constituents of biogas (CH4 and CO2) are incorporated into the final hydrocarbon product. The main catalysts of Ni, Pd, Pt supported on MgO and MgO–Ce2O3 with cubic structures that were synthesized using the co-precipitation method with K2CO3 as the precipitant has yielded good CO2 and CH4 conversion rates of 99% and 80%, respectively, for DRM at 900 °C. They also showed good thermal stability for the first 200 h. The supported catalysts, Pt, Pd, Ni/MgO and Pt, Pd, Ni/MgO–Ce2O3, with cubic structures showed good activity and thermal stability for the types of dry reforming of methane and the partial oxidation of methane reactions. These two steps occur alternately during the reforming process of the methane with the catalysts: Pt, Pd, Ni/MgO and Pt, Pd, Ni/MgO–Ce2O3. This occurrence ensures the deposition of fixed amounts of carbon on the surfaces of the catalysts, which does not affect the activity or thermal stability of the catalysts. These supported catalysts, Pt, Pd, and Ni, with cubic structures have been proven to have great potential for use in fuel processing.
Acknowledgements
The authors are thankful of NanoMite Grant (Vot. No: 5526308) for providing the funding to conduct this study. One of the authors also grateful to Basra University, Iraq for giving the financial assistance during his PhD study.
References
- K. Sutthiumporn, T. Maneerung, Y. Kathiraser and S. Kawi, Int. J. Hydrogen Energy, 2012, 37, 11195–11207 CrossRef CAS.
- M. R. Rahimpour, Z. A. Aboosadi and A. H. Jahanmiri, Appl. Energy, 2011, 88, 2691–2701 CrossRef.
- W. H. Chen, B. J. Lin, H. M. Lee and M. H. Huang, Appl. Energy, 2012, 98, 92–101 CrossRef CAS.
- M. Sarkari, F. Fazlollahi, H. Ajamein, H. Atashi, W. C. Hecker and L. L. Baxter, Fuel Process. Technol., 2014, 127, 163–170 CrossRef CAS.
- E. R. Monazam, R. W. Breault and R. Siriwardane, Chem. Eng. J., 2014, 242, 204–210 CrossRef CAS.
- A. Hasanbeigi, M. Arens and L. Price, Renewable Sustainable Energy Rev., 2014, 33, 645–658 CrossRef CAS.
- Catal. Sci. Technol, ed. J. R. Rostrup-Nielsen, J. R. Anderson and M. Boudart, Springer, New York, 1984, vol. 5 Search PubMed.
- E. Ruckenstein and Y. H. Hu, Appl. Catal., A, 1995, 133, 149–161 CrossRef CAS.
- D. Dissanayake, M. P. Rosynek and L. H. Lunsford, J. Phys. Chem., 1993, 97, 3644–3646 CrossRef CAS.
- A. T. Ashcroft, A. K. Cheetham, M. H. Green and P. F. Vernon, Nature, 1991, 352, 225–231 CrossRef CAS.
- Q. Chen, J. Zhang, Q. Jin, B. Pan, W. Kong, T. Zhao and Y. Sun, Catal. Today, 2013, 215, 251–259 CrossRef CAS.
- J. Ashok and S. Kawi, Int. J. Hydrogen Energy, 2013, 38, 13938–13949 CrossRef CAS.
- Y. Liu, Z. He, L. Zhou, Z. Hou and W. Eli, Catal. Commun., 2013, 42, 40–44 CrossRef CAS.
- Q. Chen, J. Zhang, Q. Jin, B. Pan, W. Kong, T. Zhao and Y. Sun, Catal. Today, 2013, 215, 251–259 CrossRef CAS.
- J. Kehres, J. G. Jakobsen, J. W. Andreasen, J. B. Wagner, H. Liu, A. Molenbroek and T. Vegge, J. Phys. Chem. C, 2012, 116, 21407–21415 CAS.
- M. Garcia-Dieguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, Catal. Today, 2011, 172, 136–142 CrossRef CAS.
- F. Menegazzo, M. Signoretto, P. Canton and N. Pernicone, Appl. Catal., A, 2012, 439, 80–87 CrossRef.
- E. Ruckenstein and Y. H. Hu, Chem. Innovation, 2000, 30, 39–43 CAS.
- M. Yu, K. Zhu, Z. Liu, H. Xiao, W. Deng and X. Zhou, Appl. Catal., B, 2014, 148, 177–190 CrossRef.
- K. Tomishige, Catal. Today, 2004, 89, 405–418 CrossRef CAS.
- F. W. Aldbea, N. Ibrahim, M. H. Abdullah and R. E. Shaiboub, J. Sol-Gel Sci. Technol., 2012, 62, 483–489 CrossRef CAS.
- P. Grange, Catal. Rev.: Sci. Eng., 1980, 21, 135–181 CAS.
- H. Abimanyu, C. S. Kim, B. S. Ahn and K. S. Yoo, Catal. Lett., 2007, 118, 30–35 CrossRef CAS.
- X. Chen, J. Jiang, S. Tian and K. Li, Catal. Sci. Technol., 2015, 5, 860–868 CAS.
- M. Zhiijian, L. Ying, F. Maohong and Z. Ling, J. Chem. Eng., 2015, 259, 293–302 CrossRef.
- C. Hidalgo, S. Jalila, M. Alberto, M. Jose and S. Said, J. Colloid Interface Sci., 2012, 382, 67–73 CrossRef PubMed.
- E. G. Mahoney, J. M. Pusel, S. M. Stagg-Williams and S. Faraji, J. CO2 Util., 2014, 6, 40–44 CrossRef CAS.
- Z. Bao, Y. Lu, J. Han, Y. Li and F. Yu, Appl. Catal., A, 2015, 491, 116–126 CrossRef CAS.
- C. G. Rotaru, G. Postole, M. Florea, F. Matei-Rutkovska, V. I. Pârvulescu and P. Gelin, Appl. Catal., A, 2015, 494, 29–34 CrossRef CAS.
- S. Tada, T. Shimizu, H. Kameyama and T. Haneda, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- V. M. Gonzalez-Delacruz, F. Ternero, R. Peren, A. Caballero and J. P. Holgado, Appl. Catal., A, 2010, 384, 1–9 CrossRef CAS.
- K. Y. Koo, H. S. Roh, Y. T. Seo, D. J. Seo, W. L. Yoon and S. B. Park, Int. J. Hydrogen Energy, 2008, 33, 2036–2043 CrossRef CAS.
- Z. Mei, Y. Li, M. Fan, L. Zhao and J. Zhao, Chem. Eng. J., 2015, 259, 293–302 CrossRef CAS.
- A. Djaidja, S. Libs, A. Kiennemann and A. Barama, Catal. Today, 2006, 113, 194–200 CrossRef CAS.
- H. W. Kim, K. M. Kang and H. Kwak, Int. J. Hydrogen Energy, 2009, 34, 3351–3359 CrossRef CAS.
- L. Chen, Q. Zhu, Z. Hao, T. Zhang and Z. Xie, Int. J. Hydrogen Energy, 2010, 35, 8494–8502 CrossRef CAS.
- Z. Mojovic, S. Mentus and Z. Tesic, Mater. Sci. Forum, 2004, 453, 257–262 CrossRef.
- A. M. Gadalla and M. E. Sommer, Chem. Eng. Sci., 1989, 44, 2825–2829 CrossRef CAS.
- A. Zecchina, G. Spoto, S. Coluccia and E. Guglielminotti, J. Chem. Soc., Faraday Trans., 1984, 80, 1891–1901 RSC.
- Y. H. Hu and E. Ruckenstein, Acc. Chem. Res., 2003, 36, 791–797 CrossRef CAS PubMed.
- S. Appari, V. M. Janardhanan, R. Bauri, S. Jayanti and O. Deutschmann, Appl. Catal., A, 2014, 471, 118–125 CrossRef CAS.
- P. Djinović, G. Osojnik, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- A. Topalidis, D. E. Petrakis, A. Ladavos, L. Loakatzikou and P. J. Pomonis, Catal. Today, 2007, 127, 238–245 CrossRef CAS.
- K. Nakagawa, M. Kikuchi, M. Nishitani-Gamo, H. Oda, H. Gamo, K. Ogawa and T. Ando, Energy Fuels, 2008, 22, 3566–3570 CrossRef CAS.
- T. Osaki and T. Mori, J. Catal., 2001, 204, 89–97 CrossRef CAS.
- M. M. Barroso-Quiroga and A. E. Castro-Luna, Int. J. Hydrogen Energy, 2010, 35, 6052–6056 CrossRef CAS.
- F. Giordano, A. Trovarelli, C. Leitenburg and M. Giona, Catalysis, 2000, 193, 273–282 CrossRef CAS.
- B. Steinhauer, M. R. Kasireddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333–341 CrossRef CAS.
- S. R. Miguel, I. M. J. Vilella, S. P. Maina, D. S. Jose-Alonso, M. C. Roman-Martinez and M. J. Illan-Gomez, Appl. Catal., A, 2012, 435, 10–18 CrossRef.
- J. Zhu, X. Peng, L. Yao, J. Shen, D. Tong and C. Hu, Int. J. Hydrogen Energy, 2011, 36, 7094–7104 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.