
Influence of oxodiperoxovanadate complexes on prion neuropeptide fibril formation†
Abstract
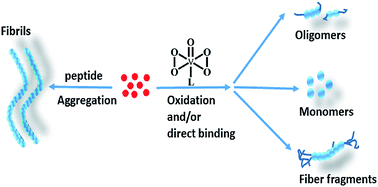
The neuropeptide PrP106–126 is used as a common model to study abnormal prion proteins. Some metal complexes are able to inhibit the formation of PrP106–126 fibrils and various compounds exhibit diverse interaction mechanisms. In this study, four oxodiperoxovanadate complexes were examined for inhibition of fibril formation of PrP106–126 and the mutant peptide M109F. Compounds 1, 3, and 4 directly interacted with the peptide and induced methionine oxidation. However, complex 2, which contains a large aromatic ligand, eliminated methionine oxidation activity and altered the inhibitory mechanism via steric effects. Aggregation was assessed by thioflavine T fluorescence and morphology analyses and complexes 1, 3, and 4 significantly decreased peptide fibril formation in comparison to 2. However, 2 caused increased cellular viability against amyloid peptide-induced cytotoxicity. This work elucidated the distinct role of a large aromatic ligand in peptide aggregation, binding affinity, and cytotoxicity.
 Please wait while we load your content...
Please wait while we load your content...

