
Layered double hydroxide and sulindac coiled and scrolled nanoassemblies for storage and drug release†‡
Abstract
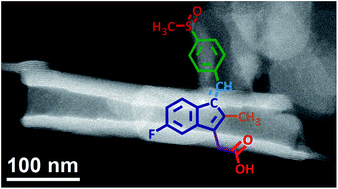
Sulindac, a non-steroidal anti-inflammatory of the indene acetic acid class, was immobilized inside layered double hydroxide Mg2Al and Zn2Al (LDH) nanovessels through a one pot reaction. LDH–drug materials were characterized by chemical elemental analysis, X-ray diffraction (XRD) (one dimensional electron distribution along the c-stacking axis, pair distribution function analysis), scanning and transmission electron microscopies, mass coupled thermal analyses, vibrational infrared and Raman spectroscopies, and solid state 13C NMR. Density Functional Theory (DFT) calculations were performed for sulindac (protonated and deprotonated forms) with the aim to assign the LDH–drug spectroscopic results. All converge towards a spatial organization of interleaved sulindac molecules close to that reported for the pristine polymorph II crystal structure. Of relevance for human treatment because of its biocompatibility and non-immunogenic effect, in vitro sulindac release experiments were performed in a phosphate buffer mimicking biological fluids and release profiles were refined using kinetic models.
 Please wait while we load your content...
Please wait while we load your content...

