
Amide-bridged terphenyl and dithienylbenzene units for semiconducting polymers†
Abstract
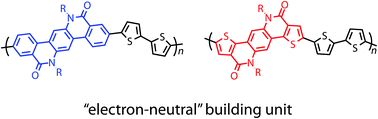
We here describe the synthesis, characterization, and structures of new semiconducting polymers (PIQP2T and PTPQ2T) based on amide-bridged terphenyl (IQP) and dithienylbenzene (TPQ), and their performances in organic field-effect transistors (OFETs) and organic photovoltaics (OPVs). The polymers are found to have relatively wide band gaps of >2.0 eV and deep HOMO energy levels of −5.4 eV. Interestingly both the HOMO and LUMO energy levels similarly shift upward and downward, respectively, by the π-extension from the monomer unit to the polymer, which can be ascribed to the delocalized HOMO and LUMO along the molecular frameworks. This suggests that IQP and TPQ can be viewed as electron-neutral building units. Both polymers had similar ordering structures in the thin film despite the fact that the IQP is more sterically hindered at the end of the moiety than TPQ. This is probably due to the strong intermolecular interactions originating in the amide group. The polymers exhibited similar hole mobilities of 0.03–0.04 cm2 V−1 s−1 in the OFET devices. Although the PCEs were modest, the OPV devices based on these polymers showed a quite high VOC of 0.94 V.
 Please wait while we load your content...
Please wait while we load your content...

