DOI:
10.1039/C5RA25758A
(Paper)
RSC Adv., 2016,
6, 12837-12849
Soy protein-modified waterborne polyurethane biocomposites with improved functionality
Received
3rd December 2015
, Accepted 20th January 2016
First published on 25th January 2016
Abstract
A series of soy protein isolate (SPI)-modified waterborne polyurethane (WPU) composite films were fabricated from WPU and SPI by a process involving blending, solution casting, and evaporation. The effects of SPI content on the structure and physical properties of the composite films were characterized by Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), thermogravimetric analysis (TGA), differential thermogravimetry (DTG), scanning electron microscopy (SEM), water absorption measurement, water contact angle measurement, and tensile testing. The results showed that the water absorption, hydrophilicity, and tensile strength in the dry state of the composite films increased with an increase of SPI content. The effects of SPI content on the cytocompatibility and biodegradability of the composite films were investigated by in vitro cell culture and in vivo implantation experiments. Cell culture experiments revealed that the composite films exhibited improved cytocompatibility and SPI promoted the adhesion, growth, and proliferation of cells. The in vivo implantation experiments illustrated that the composite films had improved biodegradability after modification with SPI. These results indicated that SPI-modified WPU composite films had great potential as biomaterials.
Introduction
With the development of materials science and engineering science, there is increased interest in the use of tissue engineered scaffolds to repair tissue injuries.1 As biological substitutes, tissue engineered scaffolds should possess appropriate mechanical properties, biodegradability, and biocompatibility.2 Previous research has revealed that some natural polymers and synthetic materials including collagen, chitosan, fibroin, polylactic acid, polycaprolactone, and polyglycolic acid could be used as tissue engineered scaffolds.3–6 However, the mechanical properties of some natural polymers are poor and the degradation products of some synthetic materials (e.g., polylactic acid) are acid, which would lead to physical discomfort and a decrease in mechanical properties.7,8
Among numerous biomedical polymers, waterborne polyurethane (WPU) could be an alternative choice. WPU made using water as solvent or dispersant is quite versatile and eco-friendly polymeric materials.9 WPU retains the excellent flexibility of polyurethanes, but is also low toxic, non-polluting, low cost, and has good applicability and safety,10 properties that are highly appreciated in the field of tissue engineering;2 however, it has been reported that WPU displayed slight cell toxicity and slow degradation rate.11–14 In view of the extensive application as a tissue engineered scaffold, the cytocompatibility and biodegradability of WPU should be enhanced. The performance of cells attached to materials is mainly affected by the surface hydrophilicity/hydrophobicity, roughness, and microstructure of the materials.15 The degradation of WPU depends on many factors such as chemical composition, micro-structure, crystallinity, crosslinking, and hard to soft segment ratio in the molecular chain.16 Three methods of modification that consider these factors have been used to enhance the cytocompatibility and biodegradability. The first method is to prepare WPU without addition of catalysts,17 thereby avoiding the potential toxicity of the catalysts to cells and improving cytocompatibility. The second method used polyester as the soft segment instead of polyether18,19 because most of the traditional petroleum-based polyethers are non-biodegradable.20 Polyester diol-based WPU films had higher tensile strength,21 therefore numerous studies on degradable waterborne polyurethanes focused on selecting polyester polyols instead of polyethers as the soft segment.22,23 The third method used natural polymers (e.g., cellulose,24 starch,25 chitosan26, and soy-based products27,28) to modify WPU via blending or chemical cross-linking. Blending methods attract more attention because of their operational simplicity compared to chemical cross-linking methods. Meanwhile, many reports have proven that a wide range of modifications to the properties of WPU films could be achieved through blending due to the synergistic effects between WPU and natural polymers.29,30 Gao et al.24 revealed that uniform distribution of cellulose nanocrystals in the WPU matrix led to significant reinforcement of tensile strength, Young's modulus, and thermal stability due to strong hydrogen bonding between the polymer matrix and filler. Shi et al.26 indicated that WPU/chitosan biocomposite films exhibited increased tensile strength and thermal stability due to the existence of hydrogen and covalent bonds between the polyurethane and chitosan. Furthermore, WPU/chitosan biocomposite films were found to be more degradable than the polyurethane alone. Zhang et al.27 found that soy protein isolate/poly(butylene adipate)-based WPU blend films displayed improved water and mechanical properties owing to the synergistic effect of soy protein isolate (SPI) and WPU.
A comprehensive method where WPU is prepared from castor oil without the addition of catalysts and modified by SPI via a blending process may be used to modify the cytocompatibility and biodegradability of WPU simultaneously. Castor oil is a renewable and degradable polyester polyol with an average functionality of 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 that reacts easily with different diisocyanates to significantly improve the flexibility, and thermal stability of WPU.32 SPI is an abundant soy-based product with high thermal stability and good biocompatibility and biodegradability33,34 that helps to mediate cell adhesion and growth.35 Blend films containing soy protein exhibit good biocompatibility, are capable of supporting cell adhesion and proliferation.30 In addition, SPI-based blend materials also display good physical or biological properties.36–39 For examples, Yang et al.36 reported that bio-based poly(lactic acid) (PLA) and soy protein blends were a compatible system and showed improved thermal stability and biodegradability. Xie et al.38 revealed that polyurethane (PU)-coated SPI films showed enhanced mechanical properties. It was found that SPI/PU composite materials with 65 wt% SPI content possessed good tensile strength and biodegradability.39 As a result, materials modified by SPI may have higher strength and biodegradability performance than that un-modified ones.40 At the same time, from the view of biomaterials, SPI and its hydrolysates can promote the cell proliferation and can be biodegraded in vitro or in vivo.41,42 Thus, it is hypothesized that SPI might be used to modify WPU to fabricate composite films which would retain the good cytocompatibility of SPI and relatively high flexibility of WPU, but have a much higher hydrophilicity and in vivo biodegradability.
31 that reacts easily with different diisocyanates to significantly improve the flexibility, and thermal stability of WPU.32 SPI is an abundant soy-based product with high thermal stability and good biocompatibility and biodegradability33,34 that helps to mediate cell adhesion and growth.35 Blend films containing soy protein exhibit good biocompatibility, are capable of supporting cell adhesion and proliferation.30 In addition, SPI-based blend materials also display good physical or biological properties.36–39 For examples, Yang et al.36 reported that bio-based poly(lactic acid) (PLA) and soy protein blends were a compatible system and showed improved thermal stability and biodegradability. Xie et al.38 revealed that polyurethane (PU)-coated SPI films showed enhanced mechanical properties. It was found that SPI/PU composite materials with 65 wt% SPI content possessed good tensile strength and biodegradability.39 As a result, materials modified by SPI may have higher strength and biodegradability performance than that un-modified ones.40 At the same time, from the view of biomaterials, SPI and its hydrolysates can promote the cell proliferation and can be biodegraded in vitro or in vivo.41,42 Thus, it is hypothesized that SPI might be used to modify WPU to fabricate composite films which would retain the good cytocompatibility of SPI and relatively high flexibility of WPU, but have a much higher hydrophilicity and in vivo biodegradability.
In this work, we prepared WPU from castor oil and hexamethylene diisocyanate without addition of catalyst, and then blend it with SPI to obtain SPI-modified WPU films. Effects of SPI content on the structure and physical properties of the SPI-modified WPU composite films were investigated by Fourier transform infrared spectroscopy, scanning electron microscopy, X-ray diffraction, thermogravimetric analysis, differential thermo-gravimetry, water absorption measurement, water contact angle measurement, and tensile testing. The cytocompatibility and biodegradability of the modified films were evaluated by in vitro cell culture and in vivo implantation experiments. SPI-modified castor oil-based WPU films were expected to possess the performance combined the excellent mechanical properties of castor oil-based WPU with the hydrophilicity, cytocompatibility, and biodegradability of SPI and to meet the requirements of tissue engineering. Therefore, these composite films are strong candidates for tissue engineering scaffolds, such as ligament and tendon scaffolds.
Materials and methods
Materials
Castor oil, from Aladdin Industrial Corporation (Shanghai, China), was vacuum dried at 60 °C for 4 h before use. Hexamethylene diisocyanate (HDI) was obtained from Aladdin Industrial Corporation and used without further treatment. Dimethylol propionic acid (DMPA) provided by Aladdin Industrial Corporation was dried at 120 °C for 4 h before use. Triethylamine (TEA), ethylic acid and ethanol were purchased from Guoyao Chemical Company (China, Shanghai). Soy protein isolate (SPI) with a protein content of 95% was purchased from DuPont Zhengzhou Protein Co. LTD (Zhengzhou, China). SPI was vacuum dried for 24 h at 60 °C before use. All other chemicals and reagents used were analytical grade.
Synthesis of WPU aqueous dispersion and SPI-modified WPU composite films
Waterborne polyurethane (WPU) aqueous dispersion was synthesized according to the process shown in Scheme 1.43 In brief: hexamethylene diisocyanate (HDI, 5.04 g) and castor oil (9.32 g) were introduced into a four-necked flask equipped with a thermometer, electric stirrer, dropping funnel, and condenser. The mixture was stirred at 70 °C for 2 h, and then dimethylol propionic acid (DMPA, 1.07 g) was added. The chain extension reaction was conducted at 85 °C for 3 h. During the period, acetone was added to reduce the viscosity of the prepolymer. The mixture was then cooled to 40–50 °C, and triethylamine (TEA, 0.8 g) was added to neutralize the carboxylic groups of DMPA. Finally, distilled water (163 g) was added to the flask under vigorous stirring to emulsify and obtain a stable 10.0 wt% WPU aqueous dispersion.
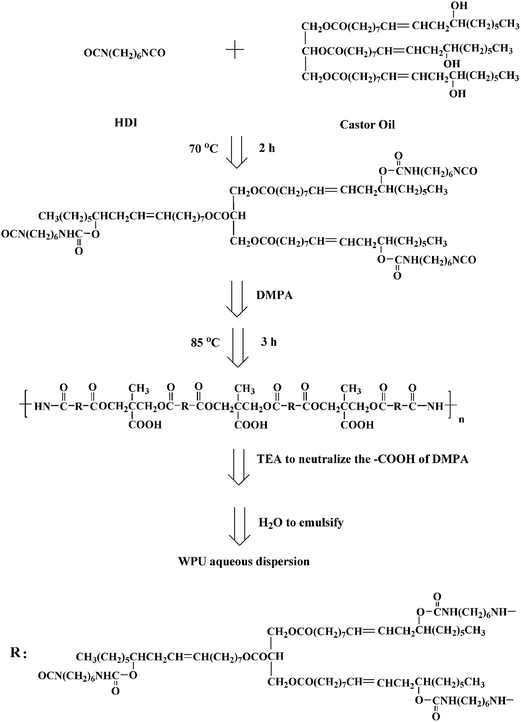 |
| | Scheme 1 Preparation of WPU aqueous dispersion. | |
Scheme 2 illustrated the synthesis route of SPI-modified WPU composite films. SPI powder was stirred in water for 10 min to obtain a slurry, and the pH of the slurry was adjusted to 10.0 with 0.5 mol L−1 NaOH solution. Then the SPI slurry was added to 10.0 wt% WPU aqueous dispersion (WPU dispersed in the form of spherical particles proven by Wang et al.,44 Peng et al.,45 and Fu et al.46) and stirred at room temperature for 30 min. After centrifuge degassing, the homogeneous composite solution was poured onto a polytetrafluoroethylene (PTFE) board at room temperature to evaporate and to obtain an even composite film. The cast composite film was immersed in 0.5 mol L−1 acetic acid solution for 30 min to neutralize NaOH and then rinsed with distilled water. Finally, composite film was dried at room temperature. A series of SPI-modified WPU composite films was fabricated and coded as WS-n, where “n” indicated the dry weight content of SPI in the composite films. For example, WS-10 means the WPU/SPI composite film contains 10 wt% SPI. Films prepared from neat waterborne polyurethane or neat soy protein isolate were coded as WPU and SPI, respectively. The codes and components of the composite films are list in Table 1.
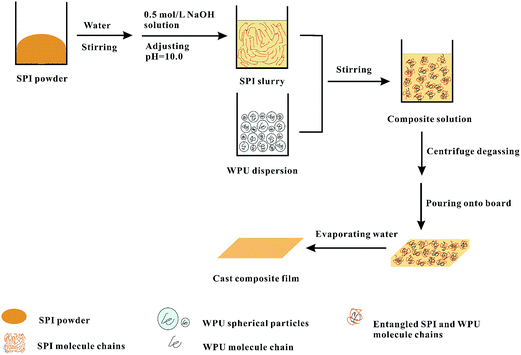 |
| | Scheme 2 Illustration of the synthesis route of SPI-modified WPU composite film. | |
Table 1 Sample codes, components, and decomposition temperatures of WPU, SPI, and WS-n composite films
| Code |
WPU content (wt%) |
SPI content (wt%) |
Temperature of 30% weight loss (°C) |
Tmax1a (°C) |
Tmax2a (°C) |
| Tmax1 and Tmax2 are the temperature at which the degradation rate of the first and the second weight loss process are maximal, respectively. |
| WPU |
100 |
0 |
290.3 |
296.7 |
445.7 |
| WS-10 |
90 |
10 |
299.1 |
328.5 |
444.3 |
| WS-30 |
70 |
30 |
292.2 |
320.1 |
439.4 |
| WS-50 |
50 |
50 |
296.5 |
311.2 |
435.8 |
| WS-70 |
30 |
70 |
300.4 |
313.9 |
429.3 |
| WS-90 |
10 |
90 |
307.7 |
324.2 |
425.5 |
| SPI |
0 |
100 |
311.1 |
340.1 |
None |
Structural characterization
Fourier transform-infrared (FTIR) analysis of the films was performed on an FTIR spectroscope (TNZI-5700, Nicolet, USA). The films were cut into powder and scanned from 4000 to 400 cm−1. The powder X-ray diffraction (XRD) patterns of the dry films were recorded on a X-ray diffractometer (D8 ADVANCE, Germany) with Cu Kα radiation with λ = 0.15406 nm. XRD data were collected from 2θ = 4–50° at a scanning rate 1° min−1. Films were immersed in the water overnight to swell and then were frozen in liquid nitrogen, immediately fractured, and freeze dried. The cross-sections of the swelling samples were coated with gold for morphologic observation using a scanning electron microscope (SEM) (VEGA3, TESCAN, Czech Republic) with a 20 kV accelerated voltage.
Thermogravimetric analysis and differential thermogravimetry
Thermogravimetric (TG) analysis and differential thermogravimetry (DTG) of the dry films were carried out on a Diamond TG/DTA instrument (PerkinElmer, USA). Thermograms were acquired between 30 and 600 °C at a heating rate of 10 °C min−1. Nitrogen was used as the purge gas at a flow rate of 20 mL min−1.
Water contact angle
Water contact angle measurements were performed using a contact angle meter (OCA20 contact angle measuring system, Data Physics Instruments GmbH, Germany). The films were fixed horizontally on a movable stage, and water droplets were deposited on the surface of samples using a micro-syringe. A CCD video camera was used to record the shape of the droplet and image analysis software was used to determine the evolution of the water contact angle. Every sample was measured at least three times and the average value was calculated.
Water absorption ratio
Water absorption ratio was measured by cutting vacuum-dried films into 3 cm × 3 cm pieces, weighing (W1) them, and then immersing in distilled water for 24 h at room temperature. When the films were taken out from water, the water on the surface was blotted with filter paper and the films were re-weighed (W2). The water absorption ratio of the films was calculated using the following equation:| | |
Water adsorption ratio (%) = [(W2 − Wl)/Wl] × 100
| (1) |
Mechanical properties
The tensile strength (σb) and elongation at break (εb) of the films were measured at a tensile rate of 10 mm min−1 on a universal testing machine (CMT4104, SANS, China) according to ISO 527-2: 2012. All samples were cut into 7 cm × 1 cm pieces. The dry films were obtained by vacuum drying at 40 °C to constant weight. Humid films were obtained by storing pieces at room temperature for one week in a closed container with 75% relative humidity (provided by saturated NaCl aqueous solution). One week was sufficient for the films to reach equilibrium. The tensile strength and elongation at break of both dry and humid films was measured, respectively. Mean values were calculated from more than three specimens, and standard deviations also calculated.
Preparation of extracts from composite films
Extracts from WPU, SPI and WS-n composite films were prepared according to ISO 10993-12: 2007. The films were immersed in cell culture medium (Gibco Minimum Essential Medium (MEM) 41500, 1.0 g film per 5 mL Gibco MEM with minor modification) at 37 °C for 72 h. The extracts were stored at 4 °C and steam-sterilized before use.
Cell viability measured by MTT assay
According to ISO 10993-5 with minor modification, a cell line of mouse lung fibroblasts (L929, provided by the China Centre for Type Culture Collection, Wuhan University, Wuhan, China) was resuspended in culture medium and plated (200 μL per well) into 96-well microtiter plates at a density of 2 × 104 cells per mL. The plates were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 24 h before the medium was replaced with steam-sterilized extracts from WPU, SPI and WS-n composite films. Culture medium with similar cells only was used as a control. After 1 and 3 days of culturing, the cells were treated with 20 μL per well MTT (5 mg mL−1 in phosphate buffered saline, PBS, filtered for sterilization) to a final concentration of 0.5 mg MTT per mL, and incubated for a further 4 h at 37 °C in a humidified atmosphere of 5% CO2. Then MTT was removed and 200 μL per well dimethyl sulfoxide (DMSO) was added to dissolve the formazan crystals. The plates were placed in an incubator at 37 °C for 10 min and then at 4 °C for 15 min before the absorbance measurements. Absorbance was read on a multiwell microplate reader (Tecan GENios, Tecan Austria GmbH, Salzburg, Austria) at a test wavelength of 570 nm. Cell viability was calculated using the following equation:| | |
Cell viability (%) = (Atest/Acontrol) × 100
| (2) |
where Atest and Acontrol are the absorption values of the test and control groups, respectively.
Cell culture on the films and cell morphology observation
Films were cut into 1 cm × 1 cm × 0.2 cm pieces and rinsed thoroughly with PBS. They were steam sterilized and then transferred to the bottom of 24-well plastic culture plates. The L929 cell suspension (200 μL), with a cell density of 2 × 105 cells per mL, was added to each sample and the control group, in triplicate, and they were cultured in a 5% CO2 humidified atmosphere at 37 °C for 24 h. Films were then washed with PBS, fixed for 2 h in 2.5 wt% glutaraldehyde, and post-fixed for 2 h in 1 wt% OsO4. The films were progressively dehydrated in ethanol after rewashing with PBS and then dried in supercritical CO2. The films with cells were mounted on stubs and coated with gold for SEM observation with an accelerating voltage of 20 kV.
In vivo evaluation
Implantation experiments were carried out according to ISO 10993-6: 2007 with minor modification. The Sprague Dawley (SD) rats (3 months, 250–300 g, all female) were from the ABSL-III Center for Animal Experiment at Wuhan University and were handled in accordance with Wuhan University Animal Care Committee. The rats were anesthetized prior to the subcutaneous implantation of WPU, WS-10, WS-50, WS-90, and SPI films (2 pieces per rat) for 1, 3, 5, and 7 months. At each given time, the rats were initially anesthetized with 5% isofluorane and then maintained at 2.5% isofluorane while implanted films were removed with a small amount of surrounding tissue. Half of the harvested implants were fixed in 4% buffered formaldehyde solution for 6 h, dehydrated, embedded in wax, vertically sectioned, and stained with hematoxylineosin (HE). Histological evaluation of the HE-stained semi-thin sections (4–6 μm) was observed by optical microscope on a Leica Q600 Qwin image analyzer. The other half of the harvested implants were fixed in 2.5 wt% glutaraldehyde and PBS (pH 7.4), then post-fixed in 1 wt% OsO4, progressively dehydrated in ethanol, dried in super-critical CO2, and coated with gold for SEM observation at an accelerating voltage of 20 kV.
Statistical analysis
The results of water absorption, water contact angle, tensile strength, and elongation at break were expressed as mean ± standard deviation (SD). Statistical significance of differences was analyzed using single factor analysis of variance (ANOVA) for P < 0.05 with Tukey's post-hoc test (Origin PRO 8.0 software).
Results and discussion
Structure and thermal properties
FTIR spectra of WPU, SPI, and WS-n films are shown in Fig. 1(a). In the FTIR spectra of SPI, there was strong absorption peak at 1624 cm−1 for the amide I band, and the absorption peak at 1533 cm−1 could be attributed to the flexural vibration of –N–H and stretching vibration of –C–N–,47 which overlapped because both WPU and SPI containing –CONH groups. The characteristic peak of –NCO (2250–2275 cm−1) was not found in the FTIR spectra of WPU and WS-n films, which indicating the –NCO has been completely reacted.48,49 For WPU, the strong absorption peak at 1257 cm−1 belonged to the stretching vibration of C–O–C.50 In this work, only castor oil contained the C–O–C group, meaning that the resultant WPU molecular chains contained castor oil chain segments. The intensity of this absorption peak decreased as SPI content increased and disappeared in the neat SPI films. This confirmed that castor oil chain segments were incorporated in WPU molecular chains. The absorption at 1697 cm−1 was assigned to the stretching vibration of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,51 which red-shifted from 1692 cm−1 in WS-10 to 1625 cm−1 in WS-70. This implied that there existed intermolecular hydrogen bond between WPU and SPI chains.30,47 As shown in Scheme 2, when SPI and WPU were mixed by mechanical agitation allowing the molecular chains to remove and disperse in water. The free WPU molecular chains entangled with SPI molecular chains during the slow evaporation of water to form new hydrogen bonds between them.
O,51 which red-shifted from 1692 cm−1 in WS-10 to 1625 cm−1 in WS-70. This implied that there existed intermolecular hydrogen bond between WPU and SPI chains.30,47 As shown in Scheme 2, when SPI and WPU were mixed by mechanical agitation allowing the molecular chains to remove and disperse in water. The free WPU molecular chains entangled with SPI molecular chains during the slow evaporation of water to form new hydrogen bonds between them.
 |
| | Fig. 1 FTIR spectra (a) and X-ray diffractograms (b) of WPU, SPI, and WS-n composite films. | |
Fig. 1(b) illustrates X-ray diffractograms of WPU, SPI, and WS-n composite films. The typical diffraction peaks of SPI were observed at 9.2° and 19.2°. The increased peak intensity at 9.2° from WPU to SPI revealed that the higher SPI content lead to an increase of the crystallinity. The typical diffraction peak of neat WPU was observed at 2θ = 20.5°, which was related to the short range regular ordered structure of both the hard and the soft domains and disordered structure of amorphous phase of the WPU matrix. Peaks broadened in the composite films indicating that SPI might affect the short-range microstructural phases of both soft and hard segments of the WPU matrix due to the interfacial interactions between WPU and SPI molecules.52 The shift of the biggest peak (20.5°) from 20.2° in WS-10 to 19.6° in WS-90 also confirmed that the interactions between WPU and SPI molecules occurred as predicted by FTIR.
The microstructure of the WPU, SPI, and WS-n composite films at swelling state were evaluated by SEM as shown in Fig. 2. It was worth noting that the cross-sections of swollen films displayed a porous structure after vacuum freeze-drying. Interestingly, larger pores appeared when the SPI content increased, which might result from the lost of partial SPI. This phenomenon suggested that porous structure might be formed when these composite films were soaked in cell culture medium or implanted and in contact with body liquid. In this case, the porous structure in the films was beneficial for cells to transport nutrients and remove metabolic byproducts, in vitro or in vivo. In other words, the incorporation of SPI into WPU may facilitate and promote cell growth and tissue invasion, which would be verified by the following results of direct cell culture on films and in vivo implantation.
 |
| | Fig. 2 SEM images of the cross-sections of WPU, SPI, and WS-n composite films at swelling state. Scale: 5 μm. | |
TGA and DTG curves of WPU, SPI, and WS-n composite films are shown in Fig. 3. WPU films displayed two successive weight loss processes, Fig. 3(a), corresponding to high speed thermal degradation at lower temperature (200–380 °C) and slow speed degradation at higher temperature (380–480 °C). The first weight loss process can be ascribed to the decomposition of urethane bonds in hard segments and the second weight loss process is associated with the decomposition of polyester polyol content in the soft segments.53 Thermal stability is an important parameter for end-use product application; from room temperature to 150 °C, the majority of composite films had no obvious weight loss. This phenomenon indicated that composite films can be sterilized by autoclave at 121 °C, which is vital to biomaterials. As seen in Fig. 3(b) and Table 1, the T30% values of all composite films exceeded that of the WPU film, showing that the thermal resistance of composite films was enhanced due to molecular interaction between WPU and SPI. The Tmax1 of WPU films was 296.7 °C and that of SPI films was 340.1 °C. The Tmax1 of all films were higher than that of WPU films because SPI contents were uniformly distributed in WPU matrix acted as thermal barriers, which greatly improved the thermal stability of composite films. In terms of Tmax2, the values of WS-n films decreased slightly, which may be owing to the decrease of polyester polyol contents.
 |
| | Fig. 3 TGA (a) and DTG (b) curves of WPU, SPI, and WS-n composite films. | |
Water contact angle and water absorption measurement
The surface hydrophilicity/hydrophobicity properties of composite films were studied by measuring the water contact angle and water absorption ratio. The effects of SPI content on the water contact angle of the films are shown in Fig. 4(a). WPU showed a high water contact angle (100°) implying that WPU films were hydrophobic. The water contact angle was the lowest (54.25°) on SPI films, indicating that SPI films were hydrophilic. The water contact angle of composite films decreased with an increase in SPI content because of the water-sensitivity derived from SPI, implying that the amount of SPI in the film modulated the surface hydrophilicity/hydrophobicity of WPU. Fig. 4(b) shows the water absorption ratio of films. The red line is the theoretical water absorption ratio and the black line is the experimental water absorption ratio. In theory, the water absorption ratio of blend films containing two components would be the total percent of water absorption ratio by the two components. However, in this work, the experimental water absorption ratio was lower than the theoretical value; this phenomenon has further proved the existence of strong interaction between WPU and SPI exactly as FTIR and XRD revealed. From the black line, it can be seen that the water absorption ratio of the neat WPU film was about 25%, whereas that of the neat SPI film was about 1300%. Results from water absorption measurement indicated that SPI was a typical hydrophilic material while WPU was a hydrophobic material, which was in accordance with the results from water contact angle measurement. As the SPI content increased from 10 to 90%, the water adsorption of the composite films increased obviously from 200 to 1000%, indicating that hydrophilicity of composite films has been enhanced significantly. In general, the hydrophilicity and hydrophobicity of the material would affect the performance of cells on its surface15 and cells were apt to adhere and grow on hydrophilic surface.54,55 SPI improved the hydrophilicity of composite films thus making them fit for cell adhesion and growth. This assumption was confirmed later by the results of cell culturing on the SPI-modified WPU films.
 |
| | Fig. 4 Water contact angle and water absorption ratio of WPU, SPI, and WS-n composite films. | |
Mechanical properties
Tensile strength (σb) and elongation at break (εb) of the dry and humid films were measured and illustrated in Fig. 5. For dry films, the σb of neat WPU was very low, only 1.07 MPa, and the value for SPI film was 27.92 MPa. It indicated that WPU films were soft and dry SPI films were hard. In general, the tensile strength and biodegradability of WPU film depends on the [NCO]/[OH] ratio and crosslinker content.48,56 In this work, in order to improve the biodegradability of the films, a relatively low [NCO]/[OH] ratio (2.17) and low DMPA content (nDMPA/nHDI = 0.27) was used. As a result, the tensile strength of the resultant neat WPU was very low (1.07 MPa). The σb gradually increased as SPI content increased. It implied that SPI content enhanced the tensile strength of composite films due to the synergistic effect between WPU and SPI caused by their intermolecular hydrogen bonding. Interestingly, the σb of humid films did not exhibit an increasing trend because water also affected the mechanical properties of WPU and SPI. Compared to dry SPI films, humid SPI films were of low tensile strength. However, both dry and humid WPU films had similar tensile strengths owing to the film's strong water-resistivity. When SPI content was higher than 50%, the σb of humid films were influenced mainly by SPI, therefore, composite films became softer and tensile strength decreased as SPI content increased. Biomaterials are used in body, so the humid strength is more important. The humid strength of WS-30 reached the highest (7.26 MPa) among the WS-n films. As SPI content increased, the humid strength of WS-n films decreased quickly. This is due to the high water absorption of SPI and low tensile strength at humid or wet state. However, all the composite films contained SPI showed higher tensile strength than WPU. In previous reports, the tensile strength of photoactive polyvinyl alcohol hydrogels (PVA) containing 30 wt% PVA was only 24.1 ± 12.2 kPa,57 and the tensile strength of poly(ethylene glycol) 860 (PEG860) hydrogels was 89.5 ± 50.7 kPa,58 which are much less than that of the films reported in this work. Thus, the present WS-n films might also have their potential applications as biomaterials. In the dry state, the εb of WPU was about 331% while SPI was about 3% meaning that SPI was brittle and WPU was flexible. In both dry and humid films, the εb decreased gradually owing to the brittleness of SPI. Although the εb decreased as SPI content increased, values for WS-50, WS-70, and WS-90 were still 138, 146, and 156%, respectively, indicating that most composite films maintained the good flexibility contributed by WPU. The εb of humid composite films was higher than that of dry composite films because water can be used as a plasticizer for SPI to increase the flexibility of the composite films in a humid or wet state.47
 |
| | Fig. 5 Tensile strength (a) and elongation at break (b) of WPU, SPI, and WS-n composite films at dry and humid state. | |
MTT assay and morphological observation of cells cultured on films
Dependence of cell viability on the cell culture time (1 and 3 days) for L929 cells cultured in film extracts is shown in Fig. 6. Cell viability of WPU was lower than that of the control (P < 0.05) on the first and third day, which indicated that WPU was slight toxic to L929 cells. These results agreed with previous reports that polyurethanes may release toxic ingredients within 1–2 days in prepared extracts.12 In this case, the slight cytotoxicity might come from complex factors. For example, when WPU dispersion was prepared, TEA was added to neutralize the carboxylic groups of DMPA. The remained TEA might slowly release from WPU during cell culture process and showed slight toxicity to the cells. Compared to WPU, cell viability of the WS-n composite films increased from WS-10 to WS-90. Furthermore, the cell viability in the extracts of WS-30, WS-50, WS-70, and WS-90 showed significant differences compared with that in WPU extracts over the 1 and 3 day culture periods (P < 0.05). These results implied that SPI decreased and even covered up the slight cytotoxity of WPU due to the positive function of SPI hydrolysates to cell proliferation. On the first day, the cell viability of composite films increased gradually as SPI content increased. Furthermore, when SPI content was higher than 50%, the cell viability of composite films was higher than that of control, which proved that composite films were no longer cytotoxic when SPI content was higher than 50%, and the non-cytotoxic characteristic of the extracts of the composite films was from the covering up effect of SPI. The cell viability on SPI and WS-90 in particular was obviously higher than that of the control (P < 0.05), which was attributed to the fact that the hydrolyzed products of SPI (such as amino acids and functional active peptides) provided nutrients required for the growth and proliferation of L929 cells.41,59 These results were consistent with the conclusions of our previous work.60 On the third day, the cell viability of SPI was lower than that of WS-90 because excessive SPI hydrolysis products disrupted the nutritional balance and were not beneficial for cell growth. When the SPI content was higher than 50% in composite films, cell viability also showed an increasing trend and was higher than that of the control. Thus, the WS-50, WS-70, and WS-90 composite films prepared in this work may meet the requirements as potential tissue engineered implants.
 |
| | Fig. 6 Dependence of cell viability of L929 cells cultured in the extracts from WPU, SPI, and WS-n composite films for 1 and 3 day. (*P < 0.05, compared to the control on the same day. #P < 0.05, compared to WPU on the same day). | |
Cytocompatibility was further investigated by cell culturing directly on the films. SEM images of L929 cells cultured on the surfaces of the films for 24 h are shown in Fig. 7. Under normal culture conditions, most of the L929 cells in the control group spread very well on the surface of the plate and had a shuttle-shaped morphology. Similar to the control group, the cells on WS-50, WS-70, WS-90, and SPI films overspread the whole surface. Furthermore, the majority of cells were also shuttled-like in shape, which indicated that WS-50, WS-70, WS-90, and SPI films displayed good cytocompatibility to L929 cells due to the non-cytotoxic nature of SPI. The cell density on the surfaces of WS-10 and WPU films was less than that of the control, and most cells were round in shape, possibly resulting from the hydrophobicity and slight cytotoxicity of WPU films. Therefore, modification of WPU films was necessary. After modification with SPI, shuttle-like L929 cells overspread most composite films owing to the increased hydrophilic properties, cytocompatibility, and porous structure of these films. In this case, the hydrophilic surface benefited cell adherence; cytocompatibility benefited cell proliferation; and the porous structure benefited nutrient transport and removal of metabolic by-products.
 |
| | Fig. 7 SEM images of L929 cells cultured on the surface of WPU, SPI, and WS-n films for 24 h. Scale: 100 μm. | |
In vivo evaluation
All animals survived the implantation experiments, and the rats didn't display abnormal phenomena, such as irritability, anorexia, illness, or weight-loss.
Histological observation. Microscope photographs of select HE-stained samples are shown in Fig. 8. From the first month to the seventh month after implantation, WPU films and WS-10 composite films exhibited a slight inflammatory reaction. WPU films and WS-10 composite films were not autologous tissues and their degradation speeds were slow, therefore, they induced an immunological rejection reaction that lasted a long time. The WS-50 films were surrounded by fibrous tissue and had an inflammatory reaction in the first month but this decreased obviously by the third month, and by the fifth month, there was only a slight inflammatory reaction and new blood capillaries had generated (as shown by black circle point in Fig. 8). This indicated that WS-50 composite films had good histocompatibility as implants allowing the fibrous tissues to exchange nutrients and metabolic by-products via blood capillaries. It was apparent that the surrounding tissue did not undergo pathological changes or inflammatory reaction, which further proved that WS-50 composite films had good histocompatibility as implants. Up to the seventh month, inflammatory reaction nearly disappeared. Comparatively, the WS-90 and SPI films degraded completely in the first month after implantation and caused a slight immunological reaction.
 |
| | Fig. 8 Microscope photographs of the HE-stained WPU, WS-10, and WS-50 films (a) after subcutaneous implantation in rats for 1, 3, 5 and 7 months. Microscope photographs of the HE-stained SPI and WS-90 films (b) after subcutaneous implantation in rats for 1 month. Black circle indicates a blood vessel. Scale: 200 μm. | |
SEM observation. In the first month after implantation, the neat SPI and WS-90 films were totally degraded, thus, Fig. 9 only displays SEM images of WPU, WS-10 and WS-50 composite films implanted in rats for 1, 3, 5, and 7 months. Some tissue was exfoliated during the preparation of WPU for SEM because the smooth, compact, hydrophobic surface of the WPU films was not suitable for tissues to adhere to and surround. When the implant time was the same, the degree of degradation depended on the SPI content; for example, in the fifth month the cross-sections of WPU and WS-10 surfaces appeared even while WS-50 appeared loose. This suggested that the degree of degradation increased as SPI content increased. WPU films were difficult to degrade because cells had difficulty entering the dense WPU structure. When the SPI content was the same, the degree of degradation was determined by the length of implant time. For example, the cross-section surface of WS-50 became looser and more porous as implant time increased, meaning that WS-50 was partially degraded in the fifth month.
 |
| | Fig. 9 SEM images of WPU films, WS-10, and WS-50 composite films after subcutaneous implantation in rats for 1, 3, 5, and 7 months. Scale: 200 μm. | |
To summarize, SPI improved the histocompatibility and accelerated the degradation of composite films. After modification by SPI, although the site of the composite film implant displayed an inflammatory reaction, it decreased over time. As well, new blood capillaries were generated and more tissue surrounded the films. When the SPI degraded, the composite films became loose and porous, and it was easier for cells to enter the composite structure. Composite films tended to degrade as the length of time of implantation grew longer. The combined properties of high flexibility, adjustable hydrophilicity/hydrophobicity, and non-cytotoxicity of SPI-modified WPU films seem to fulfill the demands required for use as scaffold in tissue-engineering, for example, as ligament or tendon scaffolds.
Conclusions
SPI-modified WPU composite films were successfully prepared. SPI showed good miscibility with castor oil-based WPU due to intermolecular interactions between WPU and SPI. SPI increased the hydrophilicity, thermal stability, and tensile strength of composite films in the dry state. Certain levels of SPI content promoted adhesion, growth, and proliferation of cells, as well as the degradation of composite films as in vivo implants. Composite films possessed appropriate mechanical properties, cytocompatibility, tissue compatibility, certain biodegradability, and thereby, might be potential candidates for tissue engineering applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. NSFC 81471789 and NSFC 81171480), ‘‘Program Cai Yuanpei 2013–2015’’ (CSC No. 201304490192 and 201304490191) from China Scholarship Council, and Fundamental Research Funds for the Central Universities (Grant No. 2042014kf0253 and 2042014kf0271).
References
- M. F. A. Cutiongco, D. E. J. Anderson, M. T. Hinds and E. K. F. Yim, Acta Biomater., 2015, 25, 97–108 CrossRef CAS PubMed.
- W. L. Shao, J. X. He, F. Sang, B. Ding, C. Li, S. Z. Cui, K. J. Li, Q. M. Han and W. L. Tan, Mater. Sci. Eng., C, 2016, 58, 342–351 CrossRef CAS PubMed.
- K. Samuelsson, D. Andersson, M. Ahldén, F. H. Fu, V. Musahl and J. Karlsson, Clin. Sports Med., 2013, 32, 111–126 CrossRef PubMed.
- Y. Itani, S. Asamura, M. Matsui, Y. Tabata and N. Isogai, Plast. Reconstr. Surg., 2014, 133, 805e–813e CrossRef CAS PubMed.
- M. S. Kim and G. H. Kim, Chem. Eng. J., 2015, 279, 317–326 CrossRef CAS.
- N. Tambe, J. Di, Z. Zhang, S. Bernacki, A. EI-Shafei and M. W. King, J. Biomed. Mater. Res., Part B, 2015, 103, 1188–1197 CrossRef CAS PubMed.
- Y. F. Zuo, J. Y. Gu, Z. B. Qiao, H. Y. Tan, J. Cao and Y. H. Zhang, Int. J. Biol. Macromol., 2015, 72, 391–402 CrossRef CAS PubMed.
- A. Salam, L. A. lucia and H. Jameel, ACS Sustainable Chem. Eng., 2015, 3(3), 524–532 CrossRef CAS.
- H. J. Yoo and H. D. Kim, J. Biomed. Mater. Res., Part B, 2008, 85, 326–333 CrossRef PubMed.
- X. Jiang, K. J. Wang, M. M. Ding, J. H. Li, H. Tan, Z. G. Wang and Q. Fu, J. Mater. Sci.: Mater. Med., 2011, 22, 819–827 CrossRef CAS PubMed.
- X. Jiang, F. L. Yu, Z. G. Wang, J. H. Li, H. Tan, M. M. Ding and Q. Fu, J. Biomater. Sci., Polym. Ed., 2010, 21, 1637–1652 CrossRef CAS PubMed.
- R. L. Santos, M. M. Pithon, A. R. B. Pereira and M. T. V. Romanos, Materia-brazil, 2012, 17, 939–945 CAS.
- N. J. Song, X. Jiang, J. H. Li, Y. Pang, J. S. Li, H. Tan and Q. Fu, Chin. J. Polym. Sci., 2013, 31(10), 1451–1462 CrossRef CAS.
- X. S. Du, Y. P. Li, X. Liu, X. Wang, C. Huselstein, Y. T. Zhao, P. R. Chang and Y. Chen, J. Mater. Sci.: Mater. Med., 2014, 25, 823–833 CrossRef CAS PubMed.
- Y. L. Luo, C. H. Zhang, F. Xu and Y. S. Chen, Polym. Adv. Technol., 2012, 23, 551–557 CrossRef CAS.
- K. Krasowska, H. Janik, A. Gradys and M. Rutkowska, J. Appl. Polym. Sci., 2012, 125, 4252–4260 CrossRef CAS.
- S. A. Madbouly, Y. Xia and M. R. Kessler, Macromolecules, 2013, 46, 4606–4616 CrossRef CAS.
- S. M. Cakic, J. V. Stamenkovic, D. M. Djordjevic and L. S. Ristic, Polym. Degrad. Stab., 2007, 43, 1838–1846 Search PubMed.
- V. D. Athawale and M. A. Kulkarni, Prog. Org. Coat., 2010, 67, 44–54 CrossRef CAS.
- S. W. Zhang, J. F. Chen, D. Han, Y. Q. Feng, C. Chang, Z. L. Song and J. Zhao, J. Coat. Technol. Res., 2015, 12(3), 563–569 CrossRef CAS.
- H. Y. Wang, Y. M. Zhou, M. He and Z. Y. Dai, Colloid Polym. Sci., 2015, 293, 875–881 CAS.
- X. Liu, K. Xu, H. Cai, J. X. Su, Z. Fu, Y. Guo and M. C. Chen, Prog. Org. Coat., 2011, 72, 612–620 CrossRef CAS.
- V. D. Athawale and M. A. Kulkarni, Prog. Org. Coat., 2010, 67, 44–54 CrossRef CAS.
- Z. Z. Gao, J. Peng, T. H. Zhong, X. B. Wang and C. Yue, Carbohydr. Polym., 2012, 87, 2068–2075 CrossRef CAS.
- T. Travinskaya, Y. Savelyev and E. Mishchuk, Polym. Degrad. Stab., 2014, 101, 102–108 CrossRef CAS.
- J. H. Shi, X. Han and Y. Kelu, Text. Res. J., 2014, 84, 1174–1182 CrossRef CAS.
- M. Zhang, F. Song, X. L. Wang and Y. Z. Wang, Colloids Surf., B, 2012, 100, 16–21 CrossRef CAS PubMed.
- Y. Chen, L. N. Zhang and L. B. Du, Ind. Eng. Chem. Res., 2003, 42, 6786–6794 CrossRef CAS.
- S. J. Gao and L. N. Zhang, Macromolecules, 2001, 34, 2202–2207 CrossRef CAS.
- H. F. Tian, Y. X. Wang, L. N. Zhang, C. Y. Quan and X. Z. Zhang, Ind. Crops Prod., 2010, 32, 13–20 CrossRef CAS.
- H. A. Mohamed, B. M. Badran, A. M. Rabie and S. M. M. Morsi, Prog. Org. Coat., 2014, 77, 965–974 CrossRef CAS.
- S. Pathan and S. Ahmad, J. Mater. Chem. A, 2013, 1, 14227–14238 CAS.
- J. E. Lee and K. M. Kim, J. Appl. Polym. Sci., 2010, 118, 2257–2263 CAS.
- S. A. Madbouly and A. Lendlein, Macromol. Mater. Eng., 2012, 297, 1213–1224 CrossRef CAS.
- K. B. Chien, E. J. Chung and N. R. Shah, J. Biomater. Appl., 2014, 28(7), 1085–1096 CrossRef CAS PubMed.
- S. Z. Yang, S. A. Madbouly, J. A. Schrader, G. Srinivasan, D. Grewell, K. G. McCabe, M. R. Kessler and W. R. Graves, Green Chem., 2015, 17, 380–393 RSC.
- F. Song, D. L. Tang, X. L. Wang and Y. Z. Wang, Biomacromolecules, 2011, 12, 3369–3380 CrossRef CAS PubMed.
- D. Y. Xie, F. Song, M. Zhang, X. L. Wang and Y. Z. Wang, Ind. Crops Prod., 2014, 54, 102–108 CrossRef CAS.
- R. Deng, Y. Chen, P. Chen, L. Zhang and B. Liao, Polym. Degrad. Stab., 2006, 91, 2189–2197 CrossRef CAS.
- Z. K. Zhong and X. Z. S. Sun, Polymer, 2001, 42, 6961–6969 CrossRef CAS.
- F. Franek, O. Hohenwarter and H. Katinger, Biotechnol. Prog., 2000, 16(5), 688–692 CrossRef CAS PubMed.
- G. A. Silva, C. M. Vaz, O. P. Coutinho, A. M. Cunha and R. L. Reis, J. Mater. Sci.: Mater. Med., 2003, 14, 1055–1066 CrossRef CAS PubMed.
- C. H. Gao, X. M. Xu, J. N. Ni and Q. Zheng, Polym. Eng. Sci., 2009, 49, 162–167 CAS.
- C. Wang, Y. D. Zheng, K. Qiao, Y. J. Xie and X. T. Zhou, RSC Adv., 2015, 5, 73882–73891 RSC.
- S. J. Peng, Y. Jin, X. F. Cheng, T. B. Sun, R. Qi and B. Z. Fan, Prog. Org. Coat., 2015, 86, 1–10 CrossRef CAS.
- C. Q. Fu, X. Z. Hu, Z. Yang, L. Shen and Z. T. Zheng, Prog. Org. Coat., 2015, 84, 18–27 CrossRef CAS.
- N. G. Wang, L. N. Zhang and J. M. Gu, J. Appl. Polym. Sci., 2005, 95, 465–473 CrossRef CAS.
- Y. Kurimoto, M. Takeda, A. Koizumi, S. Yamauchi, S. Doi and Y. Tamura, Bioresour. Technol., 2000, 74, 151–157 CrossRef CAS.
- B. Ates, S. Koytepe, M. G. Karaaslan, S. Balcioglu and S. Gulgen, Int. J. Adhes. Adhes., 2014, 49, 90–96 CrossRef CAS.
- P. M. Bummer and K. Knutson, Macromolecules, 1990, 37, 4357–4362 CrossRef.
- N. Luo, D. N. Wang and S. K. Ying, Polymer, 1996, 37, 35–77 Search PubMed.
- H. C. Kuan, C. C. M. Ma, W. P. Chang, S. M. Yuen, H. H. Wu and T. M. Lee, Compos. Sci. Technol., 2005, 65, 1703–1710 CrossRef CAS.
- H. Q. Fu, Y. Wang, W. F. Chen, W. Zhou and J. Xiao, Appl. Surf. Sci., 2015, 351, 1204–1212 CrossRef CAS.
- X. J. Tong, X. L. Luo and Y. B. Li, Ind. Crops Prod., 2015, 67, 11–17 CrossRef CAS.
- J. Wei, T. Igarashi, N. Okumori, T. Igarashi, T. Maetani, B. Liu and M. Yoshinari, Biomed. Mater., 2009, 4, 045002 CrossRef PubMed.
- T. L. Smith, J. Polym. Sci., Polym. Phys. Ed., 1974, 12, 1825–1848 CrossRef CAS.
- R. H. Schmedlen, K. S. Masters and J. L. West, Biomaterials, 2002, 23, 4325–4332 CrossRef CAS PubMed.
- J. S. Temenoff, K. A. Athanasiou, R. G. Lebaron and A. G. Mikos, J. Biomed. Mater. Res., 2002, 59, 429–437 CrossRef CAS PubMed.
- S. J. Cho, S. M. Jung, M. Kang, H. S. Shin and J. H. Youk, Polymer, 2015, 69, 95–102 CrossRef CAS.
- L. H. Luo, Y. F. Zhang, X. M. Wang, Y. Wan, P. R. Chang, D. P. Anderson and Y. Chen, J. Biomater. Appl., 2010, 24, 503–526 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 










