
Electronic structure and spectral properties of aurones as visible range fluorescent probes: a DFT/TDDFT study†
Abstract
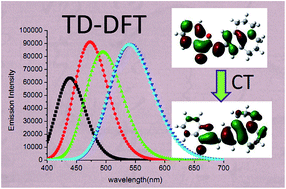
The absorption and emission spectra of aurone and its derivatives 1–4 have been investigated with density functional theory (DFT) and time-dependent density functional theory (TD-DFT). The performance of ten xc-functionals including BLYP, B3LYP, PBE0, BHHLYP, BMK, M06, M06-2X, M06-HF, LC-BLYP and CAM-B3LYP in combination with different basis sets has been analyzed. It turns out that within the selected TDDFT framework, B3LYP and PBE0 emerge as the most efficient functionals for the aurones studied. The experimentally determined spectral properties and substitution effects are well reproduced by calculations, which allowed a detailed assignment and interpretation of the spectra. The results reveal that the lowest energy transitions predominantly correspond to the π → π* transitions between the HOMO and LUMO with charge transfer (CT) character.
 Please wait while we load your content...
Please wait while we load your content...

