
An electron-transfer photochromic metal–organic framework (MOF) compound with a long-lived charge-separated state and high-contrast photoswitchable luminescence†
Abstract
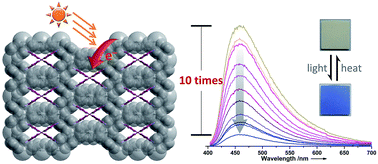
Long-lived charge-separated states and photoswitchable photoluminescence are important for many photochemical and photophysical applications. Recent studies have demonstrated the effectiveness of MOFs to achieve long-lived charge-separated states; however, the related studies are still rather rare owing to the limited numbers of photoresponsive MOFs. In this work, a new electron-transfer photochromic MOF compound, encapsulating photoactive viologen cations, was found to exhibit a charge-separated state with a lifetime value of more than 2 months, exceeding the reported values of the analogues. In addition, the photochromic process can afford a luminescence contrast up to 10 times between two stable states, which is higher than those of most known pyridine derivative-based photochromic compounds.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

