
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoluminescence and afterglow of deep red emitting SrSc2O4:Eu2+
Matthias
Müller
,
Max-Fabian
Volhard
and
Thomas
Jüstel
*
Department of Chemical Engineering, Münster University of Applied Sciences, Stegerwaldstrasse 39, 48565 Steinfurt, Germany. E-mail: tj@fh-muenster.de
First published on 21st January 2016
Abstract
This work deals with the photoluminescence (PL) properties of SrSc2O4:Eu2+ and SrSc2O4:Eu2+,Dy3+. For this purpose a series of powder samples of SrSc2O4 with various concentrations of Eu2+ and Dy3+ was prepared. The samples were synthesised via a high temperature solid state route. Phase purity was investigated by conducting X-ray powder diffractometry. The PL properties of all prepared samples were elucidated by recording PL and PL excitation spectra. Furthermore, the temperature behaviour of the PL of SrSc2O4:Eu2+ was investigated from 100 to 500 K. Diffuse reflectance spectra were recorded to investigate the optical properties of Eu2+ doped SrSc2O4. Additionally, persistent luminescence of SrSc2O4:Eu2+ and SrSc2O4:Eu2+,Dy3+ was investigated. To this end, PL lifetime measurements were conducted. SrSc2O4:Eu2+ shows an emission band in the deep red spectral range. Moreover, it turned out that the red emission of SrSc2O4:Eu2+ shows persistent luminescence too.
1. Introduction
Today, materials which show red photoluminescence (PL) or persistent luminescence are of great interest for various applications. Their uses as phosphors in light emitting diodes or as fluorescence probes in bio-imaging are only two potential fields of application for these materials. For this reason, many research groups are investigating different host compounds and various dopants to find new red emitting materials.1–3 Often, Eu2+ is taken as a dopant to obtain red emission.3,4 Since the emission of Eu2+ is due to interconfigurational d–f-transitions, the energy of the emission strongly depends on the host material. Therefore, the emission band of Eu2+ can be shifted from the ultraviolet (UV) to deep red range of the electromagnetic spectrum by changing the chemical environment.5For instance, in a material providing a surrounding with little covalent character like BaAlF5, the Eu2+ emission occurs in the UV range at about 361 nm.6 With increasing covalent interaction the emission band of Eu2+ can be located all over the visible spectrum. In CaAl2O4:Eu2+ the emission band of Eu2+ is located in the blue spectral range at about 436 nm.7 Green emission of Eu2+ can be observed in oxides and oxy-nitrides like SrAl2O4:Eu2+ (λem,max ≈ 512 nm) and β-SiAlON:Eu2+ (λem,max ≈ 535 nm). Red 5d–4f-emission of Eu2+ is commonly observed in nitrides and sulphides. For example, Sr2Si5N8:Eu2+ possesses an emission band at about 620 nm. Eu2+ doped sulfides such as CaS:Eu2+ or SrS:Eu2+ show emission at about 650 and 615 nm, respectively.
So far, not many red emitting luminescent materials on the basis of Eu2+-doped oxides are known and even less showing persistent luminescence in the deep red spectral range.1 Most of the red or infrared emitting persistent phosphors are sulphide based compounds doped with lanthanide ions.8 Especially in the red region, this group of compounds provides a variety of different emission wavelengths. Unfortunately, such compounds usually are extremely sensitive against moisture and thus chemically unstable. Therefore, further efforts like encapsulation and surface modification are necessary to protect these compounds from degradation. A chemically more stable group of compounds are oxides. Usually, oxides are impervious against moisture. However, most oxides which show red persistent luminescence are doped with Cr3+.
SrSc2O4 was firstly described and its structure solved by Carter and Feigelson in 1964.9 Later Gaume et al. reported on the spectroscopic properties of Yb3+-doped SrSc2O4.10 To the best of our knowledge there is no report concerning the PL properties of Eu2+ doped SrSc2O4. Therefore, a series of powder samples of SrSc2O4 with various contents of Eu2+ and Dy3+ was prepared. SrSc2O4 crystallizes in the orthorhombic crystal system with space group Pnam. In SrSc2O4 the Sr2+ ions are surrounded by eight O2− ions. The ionic radius of 8-fold coordinated Sr2+ is 1.26 Å. 8-Fold coordinated Eu2+and Dy3+exhibit ionic radii of 1.25 Å and 1.027 Å.11 Therefore, it is assumed that the Eu2+and Dy3+ ions tend to occupy the Sr2+ sites.
2. Experimental
The SrSc2O4:Eu2+ and SrSc2O4:Eu2+,Dy3+samples as investigated in this work were synthesized by high temperature solid state reaction. Therefore, high purity educts SrCO3 (Aldrich, 99.9%), Sc2O3 (Treibacher Industrie AG, 99.99%), Eu2O3 (Treibacher Industrie AG, 99.99%), and Dy2O3 (Treibacher Industrie AG, 99.99%) were weighted in stoichiometric amounts and were thoroughly blended in acetone in an agate mortar. After drying at ambient temperatures, the obtained powder blends were calcined at 1400 °C for 12 h in Mo boats in a reducing hydrogen atmosphere (Westfalen, 99.999%). After calcination, red sinter bodies were obtained which were ground to a fine μ-powder.Phase purity of the synthesized samples was investigated using X-ray powder diffractometry (XRD). XRD patterns were collected on a Rigaku MiniFlex II diffractometer working in Bragg–Brentano geometry using Cu Kα radiation. Step width and integration time were set to 0.02° and 1 s, respectively.
Particle size and morphology were investigated using scanning electron microscopy (SEM). Therefore, the scanning electron microscope Zeiss EVO MA10 equipped with a LaB6-cathode was used. Pressure in the sample chamber was 5 × 105 Pa and acceleration voltage was 8 kV.
PL as well as PL excitation (PLE) spectra were recorded on an Edinburgh Instruments FSL900 spectrometer equipped with a Xe arc lamp (450 W) and a cooled (−20 °C) single-photon counting photomultiplier (Hamamatsu R2658P). Obtained PL spectra were corrected by applying a correction file obtained from a tungsten incandescent lamp certified by the National Physics Laboratory U.K.
Persistent luminescence decay (PLD) curves were recorded after exciting the samples for 15 min with an excitation wavelength of λex = 525 nm. Absolute luminance and radiance was measured using the optical power meter 1830-C of Newport Corporation equipped with a Si-detector.
Temperature dependent PL measurements from 100 to 500 K were performed using the Oxford Instruments cryostat MicrostatN2. Liquid nitrogen was used as a cooling agent. Temperature stabilization time was 60 s and tolerance was set to ±3 K.
Diffuse reflectance (DR) spectra were recorded on an Edinburgh Instruments FS900 spectrometer equipped with a Xe arc lamp (450 W), a cooled (−20 °C) single-photon counting photomultiplier (Hamamatsu R928) as well as a Teflon-coated integration sphere. BaSO4 (99.998%, Sigma-Aldrich) was used as a reflectance standard.
3. Results and discussion
The collected XRD patterns as well as the ICDD reference card of SrSc2O4 are depicted in Fig. 1 for comparison purposes. The diffractograms prove the formation of the orthorhombic SrSc2O4 phase without any impurity phases. In addition, with increasing Eu2+ doping concentration no significant shift of the reflexes is observed. The diffractograms confirm the used synthesis route for the preparation of SrSc2O4:Eu2+.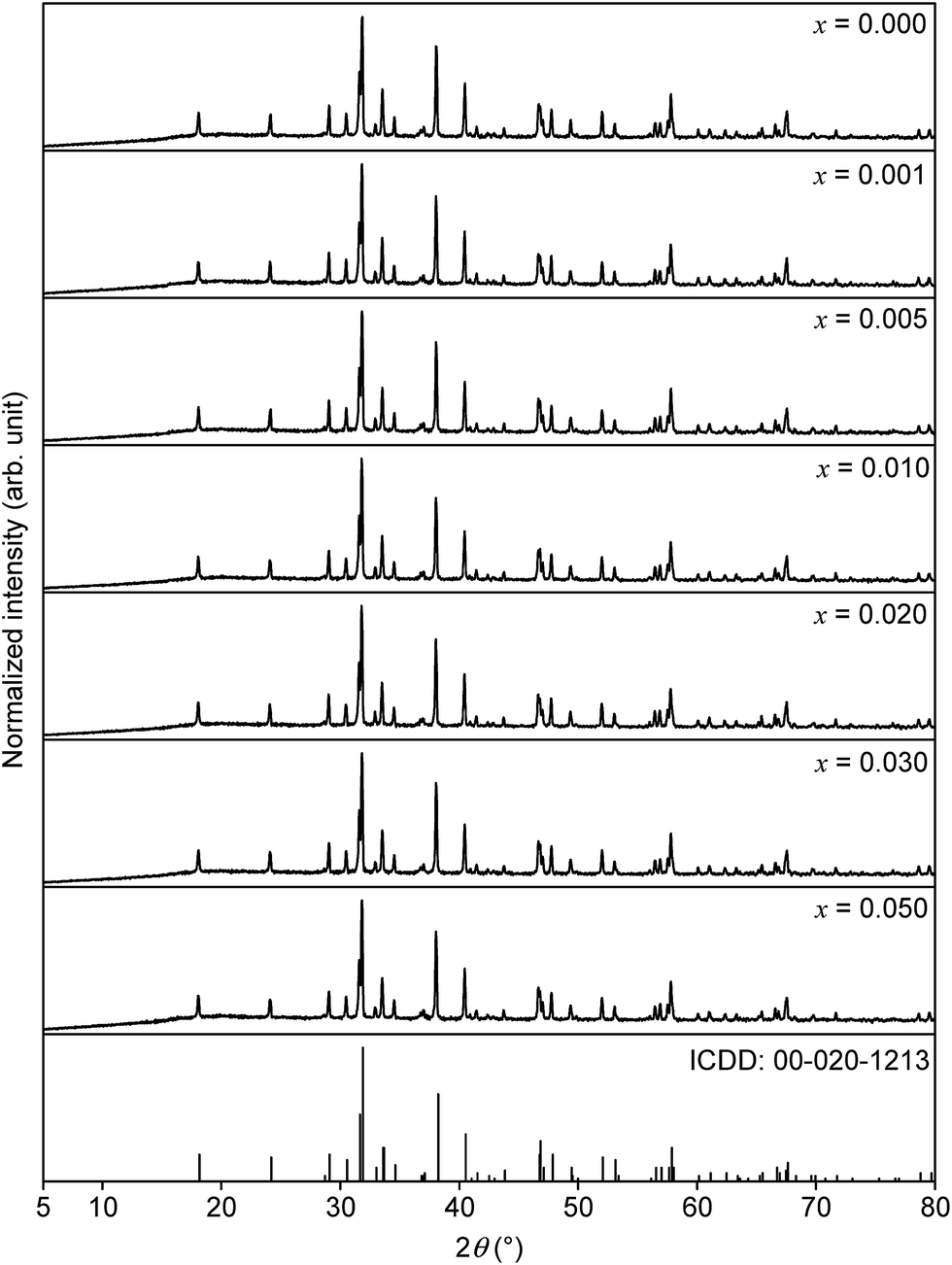 | ||
| Fig. 1 XRD patterns of Sr1−xEuxSc2O4 with various doping concentrations and ICDD reference card of SrSc2O4. | ||
Morphology of the surface as well as particle size of the prepared samples were investigated by using SEM. Fig. 2 shows the obtained image of SrSc2O4. The prepared powders of SrSc2O4 are in micrometer range. Further, the particles are heavily cracked by crushing them in the agate mortar.
 | ||
| Fig. 2 SEM photograph of SrSc2O4. | ||
Reflectance behaviour of the doped and undoped SrSc2O4 powder samples was investigated by recording DR spectra. BaSO4 was used as a reflectance standard. The obtained spectra are illustrated in Fig. 3. Undoped SrSc2O4 shows about 70% reflectance compared to BaSO4, which is in line with the white to greyish body colour. This greying is ascribed to Mo impurities caused by the Mo foil used during the high temperature annealing. The Eu2+ doped SrSc2O4 samples show strong absorption bands at about 320 and 475 nm. These bands are assigned to transitions from the 8S ([Xe]4f7) ground state to the 5d multiplet. With increasing Eu2+ concentration this absorption increases and broadens towards the long wavelength range. Therefore, the body colour of SrSc2O4:Eu2+ is shifted from light pink to red violet.
 | ||
| Fig. 3 DR spectra of Sr1−xEuxSc2O4 with various doping concentrations. | ||
PL as well as PLE spectra of Sr0.98Eu0.02Sc2O4 are depicted in Fig. 4. The PLE spectrum was recorded monitoring the Eu2+ emission at λem = 720 nm. The obtained PLE spectrum shows a broad band which consists of various unresolved bands. This band originates from the transition of the 8S ground state to the excited states 6P and 8P ([Xe]4f65d1).12 PL of SrSc2O4:Eu2+was measured upon excitation of λex = 525 nm. The obtained spectrum shows a broad band in the deep red range of the visible spectrum. This emission band ranges from about 595 to 840 nm with a maximum at about 687 nm and a full width half maximum of about 103 nm (2098 cm−1). Hence, the PL maximum of SrSc2O4:Eu2+ is located in the first bio-imaging window.
 | ||
| Fig. 4 Room temperature PL and PLE spectra of Sr0.98Eu0.02Sc2O4. | ||
The PLE spectra obtained from Sr1−xEuxSc2O4 with x = 0.02, 0.03, and 0.05 are depicted in Fig. 5. The PLE maximum shifts towards longer wavelengths with increasing Eu2+ concentration. In addition to that, the PLE spectra broaden with increasing Eu2+ concentration. The shift of the excitation maximum is mainly ascribed to a change in covalency. This is verified by calculating the centroid wavenumber of the excitation bands. The results are summarized in Table 1. With increasing Eu2+ concentration the calculated energy of the centroid wavenumbers decrease. This observation reflects an increase in the covalency of the bonds of SrSc2O4:Eu2+ and is attributed to the higher alkalinity of Eu2+ compared to Sr2+. To investigate the broadening of the excitation band in more detail, the crystal field splitting of the d-orbitals of Eu2+ was calculated (see Table 1). With increasing Eu2+ concentration crystal field splitting increases, too. Since Eu2+ is slightly smaller than Sr2+, the incorporation of Eu2+ into SrSc2O4 leads to a smaller Sr site and thus to higher crystal field splitting of the d-orbitals.
 | ||
| Fig. 5 Room temperature PLE spectra of Sr1−xEuxSc2O4 with various Eu2+ concentrations. | ||
| Sample (x) | λ ex range (nm) | Crystal field splitting (cm−1) | Centroid wavenumber (cm−1) |
|---|---|---|---|
| 0.02 | 238⋯697 | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 670 670 |
26596 |
| 0.03 | 237⋯689 | 27680 |
25381 |
| 0.05 | 235⋯685 | 27954 |
23810 |
PL spectra of Sr1−xEuxSc2O4 with x = 0.001, 0.005, 0.010, 0.020, 0.030, and 0.050 are illustrated in Fig. 6. The spectra were recorded using an excitation wavelength of λex = 525 nm. No Eu3+ emission was observed. Therefore, it is assumed that the concentration of Eu2+ corresponds to the amount which was weighed out. With increasing Eu2+ concentration the PL maximum shifts from 688 to 702 nm. In addition to that, with increasing doping concentration the PL intensity of Sr1−xEuxSc2O4 increases up to an Eu2+ content of x = 0.005. At higher concentrations the PL intensity decreases due to concentration quenching (inset Fig. 6).
 | ||
| Fig. 6 PL spectra of Sr1−xEuxSc2O4 with various Eu2+ concentrations. Inset: integrated PL intensity in dependence of the Eu2+ concentration. | ||
To investigate the mechanism of the energy transfer responsible for concentration quenching, van Uitert's approach was applied:13
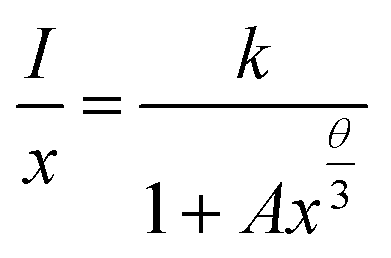 | (1) |
 | ||
| Fig. 7 Dependence of I/I0 on the Eu2+ concentration. | ||
Furthermore, the critical distance Rc between the Eu2+ ions was calculated. Rc is defined as the distance, at which the probability of energy transfer equals the probability of radiative transition. To estimate the critical distance Rc between Eu2+ ions in SrSc2O4:Eu2+ Dexter's formula for dipole–dipole interaction can be used:14
 | (2) |
In this equation QA is the absorption cross section of Eu2+ and E is the energy of maximum spectral overlap. ∫FS(E)FA(E)dE represents the spectral overlap of the normalized PL (FS(E)) and PLE (FA(E)) spectrum of Eu2+. QA is 4.8 × 10−16fd. Here, fd is the oscillator strength of the electronic transitions of Eu2+ and was taken as 0.02. The spectral overlap integral was calculated to be 0.03 eV−1. Plugging these values into eqn (2)Rc is calculated to be 20.3 Å.
In order to examine the temperature behaviour of the PL of SrSc2O4:Eu2+ PL spectra were recorded from 100 to 500 K. The resulting spectra are illustrated in Fig. 8. With increasing temperature the PL intensity gradually decreases. Moreover, the emission maximum shifts from 692 to 680 nm. Due to extending equilibrium distance between Eu2+ and its surrounding oxygen ligands with increasing temperature, crystal field splitting of the d-orbitals of Eu2+decreases. As a consequence, the energetic distance between the d and f-orbitals increases and the emission maximum is shifted towards higher energy. The inset of Fig. 8 depicts the integrated PL intensity of the investigated sample. Fitting these data points with a Fermi–Dirac distribution yields the activation energy EA for thermal quenching.
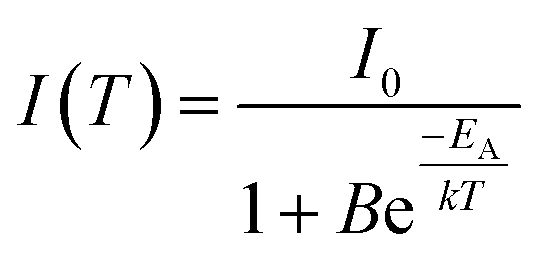 | (3) |
 | ||
| Fig. 8 PL spectra of Sr0.995Eu0.005Sc2O4 from 100 to 500 K. Inset: integrated PL intensity in dependence of the temperature. | ||
In eqn (3)I(T) is the PL intensity at a certain temperature and I0 is the PL intensity at zero kelvin. B is the frequency factor for thermal quenching, k = 8.617 × 10−5 eV K−1 is the Boltzmann constant, and T is the temperature. Parameters I0 and B were derived from the fitting function and are about 0.97 and 2945, respectively. From this EA was calculated to be 0.25 eV. By applying EA to the following formula, T1/2 can be calculated:
 | (4) |
To investigate the persistent luminescence properties of Eu2+ doped SrSc2O4, luminescent lifetime measurements were conducted. For that purpose, the investigated samples were charged for 15 min with an excitation wavelength of λex = 525 nm. Fig. 9 shows the obtained PLD curves of Sr1−xEuxSc2O4 with x = 0.001, 0.005, and 0.010. The longest afterglow duration was found for Sr0.999Eu0.001Sc2O4. The corresponding PLD curve can be best fitted with the following equation:
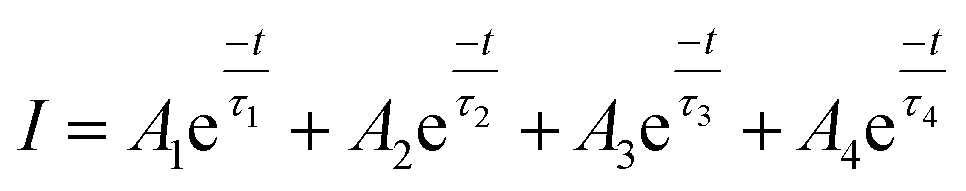 | (5) |
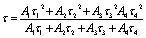 | (6) |
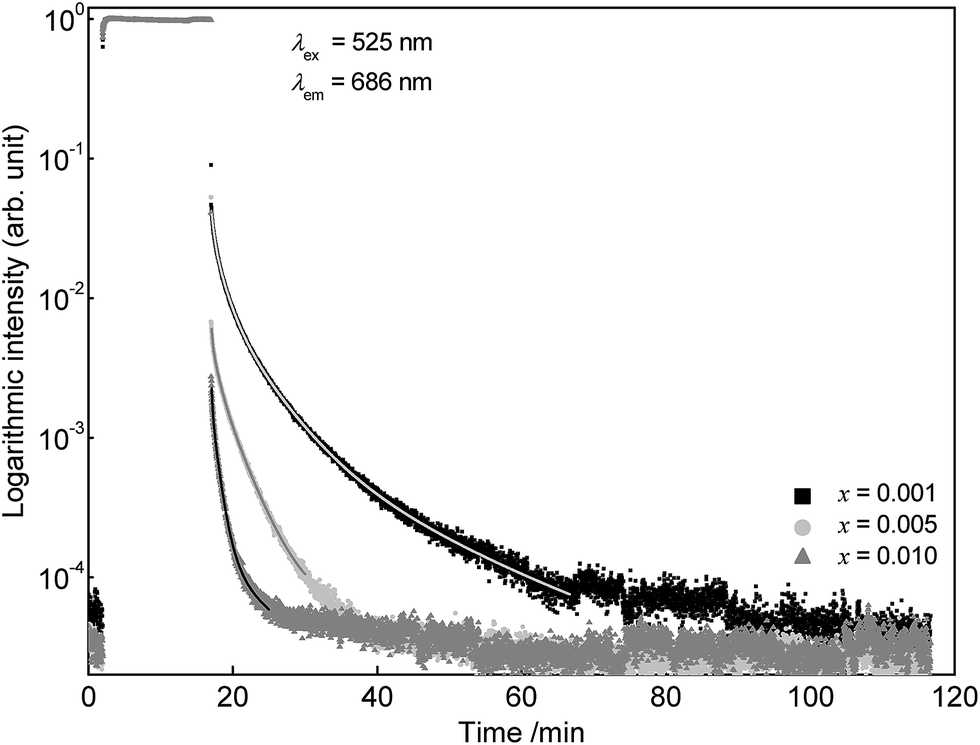 | ||
| Fig. 9 PLD curves of Sr1−xEuxSc2O4 with various doping concentrations. | ||
Partial lifetimes and their corresponding emission fractions as well as the calculated average lifetime τ are summarized in Table 2. With increasing Eu2+ content the PL lifetime of SrSc2O4:Eu2+ decreases and the PLD curve becomes tri-exponential.
| Sample (x, y) | τ 1 (s) | frac1 (%) | τ 2 (s) | frac2 (%) | τ 3 (s) | frac3 (%) | τ 4 (s) | frac4 (%) | τ (s) |
|---|---|---|---|---|---|---|---|---|---|
| 0.001, 0.000 | 21.7 | 4.8 | 88.3 | 27.4 | 293.6 | 47.3 | 1007.4 | 20.6 | 371 |
| 0.005, 0.000 | 10.4 | 2.3 | 57.9 | 18.7 | 193.6 | 79.0 | 164 | ||
| 0.010, 0.000 | 9.6 | 6.2 | 56.1 | 59.7 | 283.8 | 34.1 | 131 | ||
| 0.001, 0.001 | 16.2 | 3 | 93.7 | 32.0 | 271.1 | 65.0 | 207 | ||
| 0.001, 0.005 | 9.2 | 2.0 | 84.8 | 32.2 | 257.0 | 65.9 | 197 |
It is well known that additional incorporation of Dy3+ into Eu2+ doped materials can lead to long lasting persistent phosphors.15 In order to investigate the influence of Dy3+ co-doping on the afterglow duration, additional luminescence lifetime measurements were performed on Sr0.998Eu0.001Dy0.001Sc2O4 and Sr0.994Eu0.001Dy0.005Sc2O4. Fig. 10 illustrates the PLD curves of co-doped SrSc2O4:Eu2+,Dy3+. Analogous to Eu2+, with increasing Dy3+ concentration the afterglow duration decreases. Another possibility to enhance the afterglow duration of SrSc2O4:Eu2+ could be co-doping with Cr3+.16 Further experiments should be performed to investigate the influence of Cr3+ co-doping on the persistent luminescence properties of SrSc2O4:Eu2+.
 | ||
| Fig. 10 PLD curves of Sr0.999−yEu0.001DyySc2O4 with different concentrations of Dy3+. | ||
Fig. 11a depicts the luminance in dependence of the time. For comparison the solid line visualises the luminance value 0.32 mcd × m−2 which is the minimum value commonly used for safety signage.17 The dashed line illustrates the limit of the sensitivity of the dark-adapted eye. For the Sr0.999Eu0.001Sc2O4 sample 0.32 mcd × m−2 is reached after about 6 min. Absolute radiance is plotted in Fig. 11b. The absolute radiance values obtained after charging the sample for 15 min are considerably smaller compared to other deep red emitting persistent phosphors.18
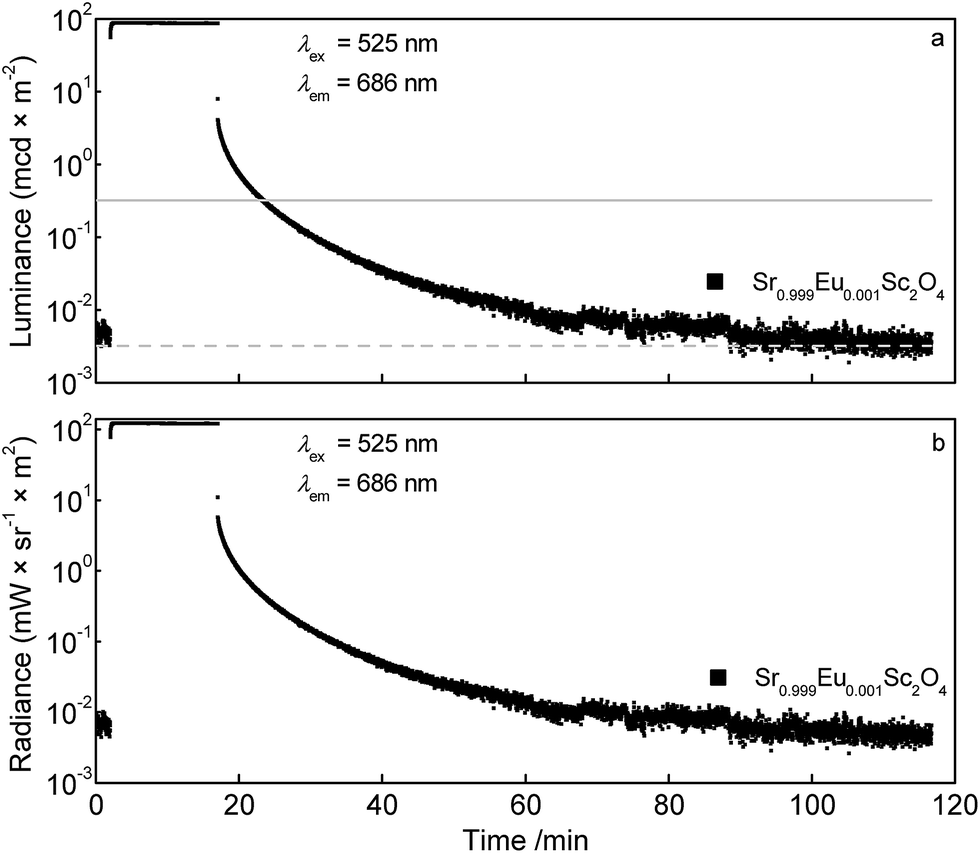 | ||
| Fig. 11 Persistent luminescence curves of Sr0.999Eu0.001Sc2O4: luminance (a) and radiance (b) of the Eu2+ emission. | ||
4. Conclusions
SrSc2O4:Eu2+ powder samples were prepared and its PL properties were investigated, also time and temperature dependent. SrSc2O4:Eu2+ shows a broad emission band in the deep red region of the spectrum with a maximum at about 687 nm. This deep red lying emission band is quite rare for oxidic host materials doped with Eu2+.1 Further, no Eu3+ emission was observed. The highest PL intensity was found for a Eu2+ concentration of x = 0.005. At a higher Eu2+ concentration the PL intensity decreases due to concentration quenching caused by energy transfer between Eu2+ ions, likely resulting in energy migration to defect states. It turned out that in SrSc2O4:Eu2+ energy transfer between Eu2+ ions is of a resonance type and occurs via dipole–dipole interaction. The critical distance Rc was calculated to be about 20.3 Å. Temperature dependent PL measurements revealed a T1/2 value of 357 K. Additionally, it was found that SrSc2O4:Eu2+ shows deep red persistent luminescence after charging it with an excitation wavelength of λex = 525 nm. The longest afterglow duration was found for the sample Sr0.999Eu0.001Sc2O4. This sample reaches the value of 0.32 mcd × m−2 after about 6 min what is considerably shorter than for comparable materials.18 No increase in afterglow duration could be observed if SrSc2O4:Eu2+ was additionally doped by Dy3+. For this reason, it would be worth the effort to try to increase the afterglow duration by doping with additional ions like Cr3+.Acknowledgements
The authors are grateful to Merck KGaA Darmstadt, Germany for generous financial support.References
- D. Deng, H. Yu, Y. Li, Y. Hua, G. Jia, S. Zhao, H. Wang, L. Huang, Y. Li, C. Li and S. Xu, J. Mater. Chem. C, 2013, 1, 3194 RSC
.
-
(a) K. Song, J. Zhang, Y. Liu, C. Zhang, J. Jiang, H. Jiang and H.-B. Qin, J. Phys. Chem. C, 2015, 119, 24558–24563 CrossRef CAS
- P. Pust, V. Weiler, C. Hecht, A. Tücks, A. S. Wochnik, A.-K. Henß, D. Wiechert, C. Scheu, P. J. Schmidt and W. Schnick, Nat. Mater., 2014, 13, 891–896 CrossRef CAS
-
(a) R.-J. Xie, N. Hirosaki, T. Suehiro, F.-F. Xu and M. Mitomo, Chem. Mater., 2006, 18, 5578–5583 CrossRef CAS
-
(a) S. H. M. Poort, H. M. Reijnhoudt, H. O. T. van der Kuip and G. Blasse, J. Alloys Compd., 1996, 241, 75–81 CrossRef CAS
- W. Zhang, R. Hua, T. Liu, J. Zhao, L. Na and B. Chen, Mater. Res. Bull., 2014, 60, 247–251 CrossRef CAS
-
(a) C. Chang, J. Xu, L. Jiang, D. Mao and W. Ying, Mater. Chem. Phys., 2006, 98, 509–513 CrossRef CAS
-
(a) J. Zhang, Y.-L. Liu and S.-q. Man, J. Lumin., 2006, 117, 141–146 CrossRef CAS
- J. R. Carter and R. S. Feigelson, J. Am. Ceram. Soc., 1964, 47, 141–144 CrossRef CAS
- R. Gaume, B. Viana, J. Derouet and D. Vivien, Opt. Mater., 2003, 22, 107–115 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
-
(a) G. Blasse, Phys. Status Solidi B, 1973, 55, K131–K134 CrossRef CAS
- L. G. van Uitert, J. Electrochem. Soc., 1967, 114, 1048 CrossRef CAS
-
(a) D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS
-
(a) I. P. Sahu, D. P. Bisen, N. Brahme and M. Ganjir, Luminescence, 2015, 30, 1318–1325 CrossRef CAS
-
(a) H. Ryu and K. S. Bartwal, J. Alloys Compd., 2008, 464, 317–321 CrossRef CAS
- K. van den Eeckhout, D. Poelman and P. Smet, Materials, 2013, 6, 2789–2818 CrossRef CAS
-
(a) J. Xu, S. Tanabe, A. D. Sontakke and J. Ueda, Appl. Phys. Lett., 2015, 107, 81903 CrossRef
| This journal is © The Royal Society of Chemistry 2016 |