
Catalytic oxidation of nitric oxide (NO) with carbonaceous materials
Abstract
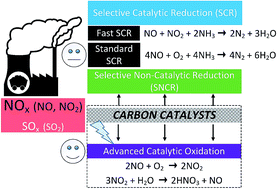
The catalytic oxidation of NO to NO2 at ambient temperatures has been a promising route for controlling NO emissions, since NO2 is subsequently removed as nitric acid in the presence of water. Because of their large surface area, high porosity, and relative chemical inertness, carbon-based materials are very attractive in de-nitrification (De-NOx) as catalysts or catalyst supports. This paper reviewed the catalytic oxidation of NO to NO2 over commonly-used carbon materials including activated carbons (ACs), activated carbon fibers (ACFs) and carbon xerogels (CXs). The NO conversion is often influenced by the surface characteristics of carbon materials (e.g., pore structure, surface areas, functional groups, and morphology), O2 concentration, and reaction temperature. With the addition of metal actives, the catalytic performance could be significantly improved. Catalytic reaction and adsorption are two key points. Further, the strong dependence of NO conversion on the O2 concentration concludes that O2 is first adsorbed on the carbon surface, and then it reacts with NO to form adsorbed NO2, which desorbs to the gas phase. Considering the economic efficiency, carbon precursors from biomasses could be fabricated into the desired carbonaceous materials by means of functionalization. In addition, the integrated strategy of desulfurization (De-SOx) and De-NOx could be developed by carbon materials with the proper modification methods.
 Please wait while we load your content...
Please wait while we load your content...

