
A time-insensitive colorimetric sensor for the determination of total protein†
Bahram Hemmateenejad*ab,
Arezoo Shahrivar-kevishahia,
Fatemeh Shakerizadeh-Shirazia,
Shohre Rouhanicd and
Fereshteh Mohamadi-Gharaghania
aDepartment of Chemistry, Shiraz University, Shiraz, Iran. E-mail: hemmatb@shirazu.ac.ir; Fax: +98 713 646 0788; Tel: +98 713 646 0724
bMedicinal and Natural Products Chemistry Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
cDepartment of Organic Colorants, Institute for Color Science and Technology, Tehran, Iran
dCenter of Excellence for Color Science and Technology, Institute for Color Science and Technology, Tehran, Iran
First published on 19th May 2016
Abstract
Quantification of protein concentration is clinically important and especially in low concentration is of great significance. UV/vis absorbance spectrometry is the routinely used measurement method for protein quantization. However, most of the reported methods tend to overestimation, and more critically, depend highly on the timing, protein variation and suffer from interference in the measurements. Here, we report a simple, time-insensitive and superior sensitive method based on the binding of a new cationic cyanine dye with BSA. Both UV/vis spectrophotometry and image analysis techniques were used as a detection system. Optimum conditions were investigated by the experimental design method. This method is more convenient because it allows for UV spectroscopy measurements in time durations of >25 min. Two linear ranges for BSA are 22.00–112.00 nM and 0.43–4.10 μM and the limit of detection is 6.25 nM. The relative standard deviation of six replicate measurements is 2.33% for 2.50 μM BSA. This method, which responds to total protein, is simple, practical, and relatively free of interference from coexisting substances. It was successfully applied to the determination of protein in urine samples.
Introduction
Proteins are one of the most fundamental groups of molecules in the human body. They are complex combinations of smaller chemical compounds called amino acids.1 Detection and quantification of total protein content is a measurement common to many applications in basic science research and routine clinical laboratory practice. Most biochemical studies that involve the measurement of a biological activity require the normalization of that activity to the protein content. The ability to easily and reliably quantitate total protein content in samples is paramount to many biological assays.Several protein assays are widely employed for protein determination.2 Many of them involve protein–dye binding chemistry (Coomassie/Bradford)3 methods or protein–copper chelation chemistry (Lowry method,4 biuret method, bicinchoninic acid (BCA) assay).5 These assays require a calibration process prior to determination of an unknown protein concentration, so if the unknown protein sample has similar characteristics to the protein measured in the standard curve, the concentration of the unknown could be determined with a high degree of accuracy. However, if the unknown protein sample has many different characteristics to the protein measured in the standard curve the error in the concentration determined may be significant.
Protein-to-protein variability, interference from contaminants, and the requirement for carefully timed reagent additions and incubations are common limitations of protein quantitation assays. Recently, fluorescent protein assays have been developed with improved sensitivity,6 but the cost per assay can make them unacceptable for large numbers of samples. Accordingly, there is great interest in developing sensitive and accurate assays of proteins in solution, which are useful over a wide range of protein concentrations especially in low concentrations.
Very recently, our research has been focused on the development of simple, inexpensive and fast bioanalytical methods. In this regard, we use image capturing device as an alternative to visible spectrophotometric equipment.7–10 Imaging method is used to perform fast, non-invasive and low-cost analysis of products and processes.
In this work, besides to spectrophotometric measurements, we employed image analysis technique for monitoring the color change of solutions as function of analyte concentration. In the other words, we use the color density of the solutions as an alternative analytical signal for the detection and determination of bovine serum albumin (BSA) as a protein representative. The color values in the RGB space were correlated with the absorbance spectra of the solutions. So, we suggested the use of color values of the images recorded by a digital camera (instead of absorbance) as a suitable analytical probe to evaluate the BSA concentration.
The selective binding of a newly synthesized cyanine dye indicator (Fig. 1) to protein was used to develop a colorimetric method for assay of proteins. The visual color change (from pink to blue) of this indicator in the presence of protein allowed sensitive determination of protein in artificial urine and blood plasma. Besides to very low detection limit, it offered high accuracy and very fast response time.
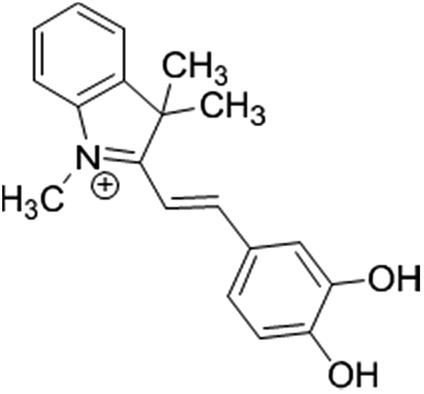 | ||
| Fig. 1 The chemical structure of the used cyanine dye. | ||
Experimental
Reagents and materials
Deionized distilled water was used throughout all experiments. Human serum albumin (fatty acid-free HSA) and bovine serum albumin (BSA) were purchased from Sigma Aldrich (St. Louis, MO, USA) and used without further purification. The spectrophotometric detection kit for protein assay based on bromocresol green reagent was provided from Pars Azmoon Company (Tehran, Iran). All other chemicals were of analytical reagent grade; sodium hydroxide (NaOH), sodium chloride (NaCl), calcium chloride (CaCl2), trisodium citrate (Na3C6H5O7), Tris-hydroxymethyl-methane (Tris), glucose, magnesium chloride (MgCl2), sodium oxalate (Na2C2O4), urea (CH4N2O), monosodium phosphate (NaH2PO4), disodium hydrogen phosphate (Na2HPO4), sodium sulfate (Na2SO4), sodium hydrogen carbonate (NaHCO3), magnesium sulfate (MgSO4), ammonium chloride (NH4Cl), uric acid (C5H4N4O3) and potassium chloride (KCl) were obtained from Merck. Creatinine was purchased from Fluka.The stock solutions of BSA or HSA were prepared by dissolving the solid protein in buffer solution (pH = 7.4) and stored at 0–4 °C in the dark for about a week only and then diluted to 1.0 × 10−6 mol L−1 using Tris–HCl buffer (pH = 7.4), when needed. The concentrations of BSA and HSA were determined from optical density measurements, using the values of molar absorptivity of ε280 = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 720 and 35700 M−1 cm−1 for BSA and HSA, respectively.11
720 and 35700 M−1 cm−1 for BSA and HSA, respectively.11
The cyanine dye (Fig. 1) was synthesized by Rouhani research group.12 Briefly, a solution of 3,4-dihydroxybenzaldehyde (0.05 mol) in ethanol, 0.05 mol 1,3,3-trimethyl-2-methylene indoline (Fischer's base) and 0.1 mol ammonium acetate as catalyst was added and after stirring for one hour, then the mixture was refluxed. The progress of the reaction mixture was monitored by TLC. After completion of reaction, the precipitate was filtered and washed with acidic water. The product was recrystallized from ethanol. This resulted in a dark and shiny green solid with 83% yield and mp 176 °C. The cyanine dye was characterized by IR, 1H NMR and mass data. The results were assigned as follows: FTIR (KBr) (νmax/cm−1): 3233, 1485, 1350, 1308, 1217, 1177, 1101; 1H NMR (500 MHz, DMSO, δ ppm): 1.75 (s, 6H), 4.07 (s, 3H), 7.04 (d, j = 8 Hz, 1H), 7.34 (d, j = 15 Hz, 1H), 7.53–7.63 (m, 3H), 7.67 (d, j = 2 Hz, 1H), 7.81–7.83 (m, 2H), 8.28 (d, j = 15 Hz, 1H); MS (m/z): 294.2. The chemical structure of this dye is shown in Fig. 1. Stock solution of dye was prepared by dissolving its crystals in minimum of HCl (1.0 mol L−1) and then proper dilution with doubly distilled water.
Apparatus
Absorption spectra were recorded on a BEL-Gold-spectrum-Lab 53 UV-vis spectrophotometer equipped with a 1 cm quartz cell. The pH measurements were carried out using a Metrohm pH-meter (model 827) with a combined pH glass electrode, calibrated against standard buffer solution of pH 4.0 and 7.0.Procedure
Results and discussion
Spectroscopy characteristics of BSA–dye complex
As it is observed from Fig. 1, the used dye possesses two ionisable hydroxyl groups. Thus, it can show pH-dependent spectral characteristics. It is clear from the ESI Fig. S2† that at acidic pH, the dye has a maximum absorbance peak at around 420 nm. However, by increasing the pH to neutral and basic, the absorbance peak maxima shifts to around 540 nm. So, to neglect the effect of pH on spectral changes, the interaction of dye with protein was conducted in buffered solution. In Fig. 2 are shown changes in the absorbance spectra of the dye interacted with BSA in Tris–HCl buffer solution of pH 7.4. It is observed that in the wavelength range of 400–750 nm, BSA shows no absorbance while cationic cyanine dye has absorption band in the visible region with a maximum at 541 nm. The absorbance of the dye in the presence of BSA decreases at around 541 nm in accompanying with a slight increase in absorbance at about 650 nm and formation of a new band. The observed bathochromic-shifted resulted in naked eye visualization of color change from pinkish-red to bluish in the presence of BSA. These variations in absorption spectra of dye after addition of BSA suggested formation of a new protein–dye complex. Previous spectrofluorometric studies revealed that the interaction of the dye with BSA is mainly derived by electrostatic interactions.12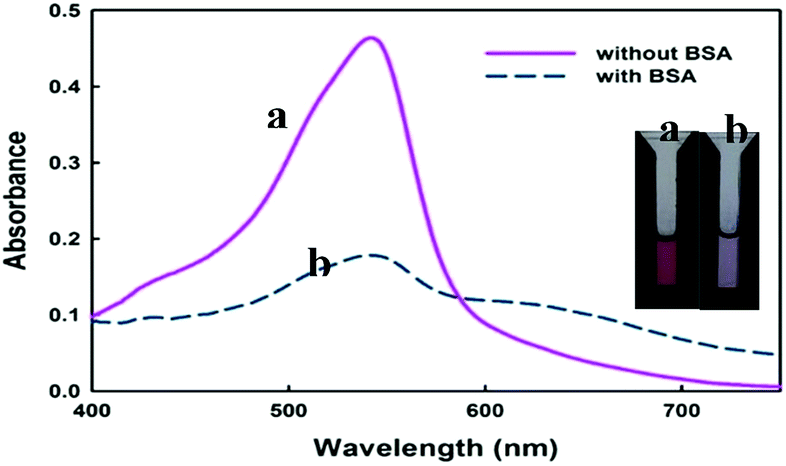 | ||
| Fig. 2 Absorbance spectra and photograph of the cyanine dye solution in the absence (a) and in the presence (b) of BSA (2.3 μM). In both solutions the pH was kept fixed at 7.4 using Tris–HCl buffer and the only difference is the presence or absence of BSA. | ||
To gain a more insight in the interaction mechanism of dye with BSA, circular dichroism (CD) spectroscopy was used. It is evident from (Fig. 3) that the CD spectra for BSA show two minima at about 208 and 222 nm, which is a clear signature of the presence of α-helix in the BSA tertiary structure. In their native forms, the percentage of α-helix in BSA has been estimated to be 60%. Interaction of dye with BSA causes increasing in α-helix content to about 63% which shows the stabilization of albumin during this interaction.
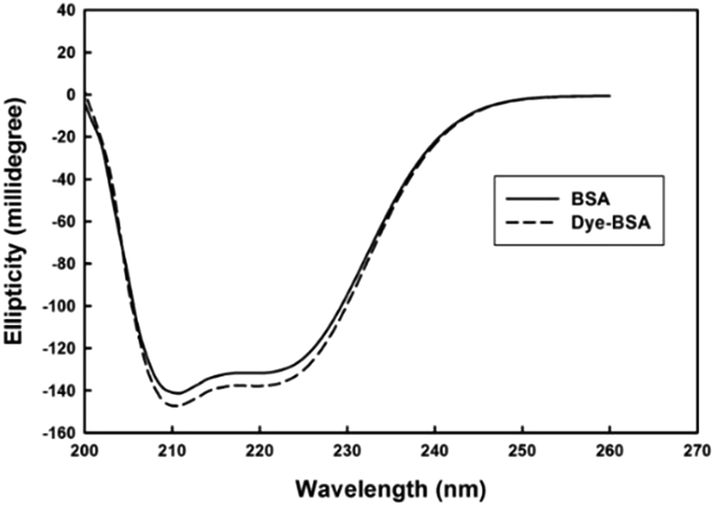 | ||
| Fig. 3 Circular dichroism spectra of BSA and dye–BSA complex. | ||
Optimization by experimental design
In the primary study of the effect of pH and dye concentration using one-at-a-time method, it was observed that the effect of these parameters on dye–protein binding was not independent. In the other words, there is interaction between parameters. Thus, to find optimum condition, experimental design13 was used. In this case, the effects of pH and dye concentration on the absorbance change were studied, simultaneously. To do so, a 3-level full factorial design was adopted. The obtained response surface is shown in Fig. 4. As seen, both factors represented a quadratic effect on the response. However, the effect of pH is more pronounced. At pH values higher than the isoelectric point of BSA (pH of 4.9), the protein is bearing negative charge. On the other hand, the dye is carrying a positive charge because of the presence of positive iminium group. Thus, by increasing in pH, more increase in negative charge of protein and stronger electrostatic interaction is resulted. However, at higher pH, the ionization of the hydroxyl groups of the dye diminishes the electrostatic interactions.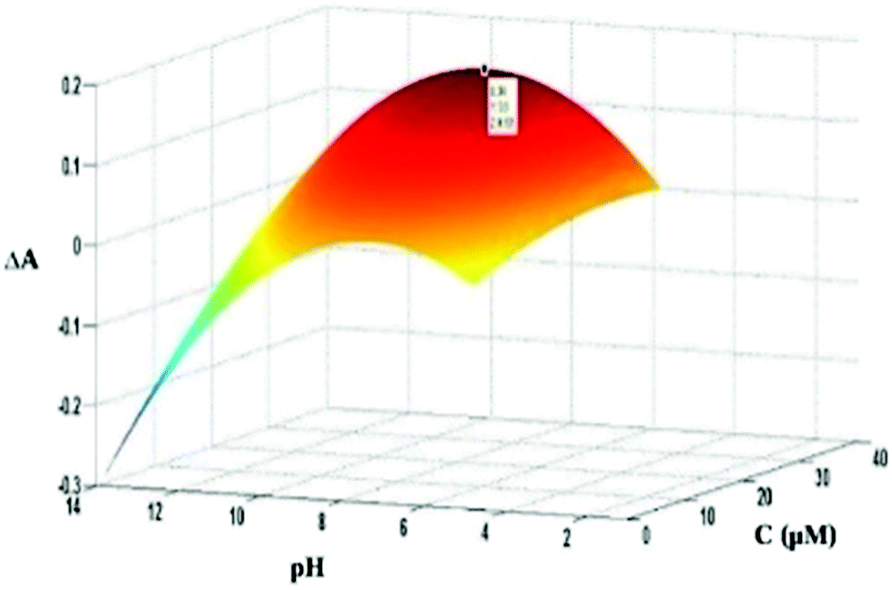 | ||
| Fig. 4 Response surface plot for investigating the effect of pH and dye concentration on absorbance changes. | ||
According to the shown response surface plot and fitting of the data to a 5-parametric model (two linear, two quadratic and one interaction terms), the optimum values of 9.3 and 34.0 μM were obtained for pH and dye concentration, respectively. However, as it is observed from (Fig. 4), there is a small difference between the response at optimum pH (9.3) and the physiological pH (7.4). So in the next analysis, both physiological pH and optimum were investigated.
Effect of the type and concentration of buffer
After finding the optimum pH, the effect of type of buffer solution was also studied. To do so, phosphate buffer and Tris–HCl buffers were used as the buffer moiety with the same pH values. Experiments at different buffer solutions showed that the dye was more responsive to BSA in Tris–HCl buffer than phosphate buffer (Fig. 5). Thus; the rest of this study were performed in Tris–HCl buffer. Also, the results indicated that buffer concentration has little effect on dye–protein, binding and at concentration of >0.05 M the effect is independent of buffer concentration (Fig. 5). Therefore, a 0.05 M buffer solution was chosen in this work.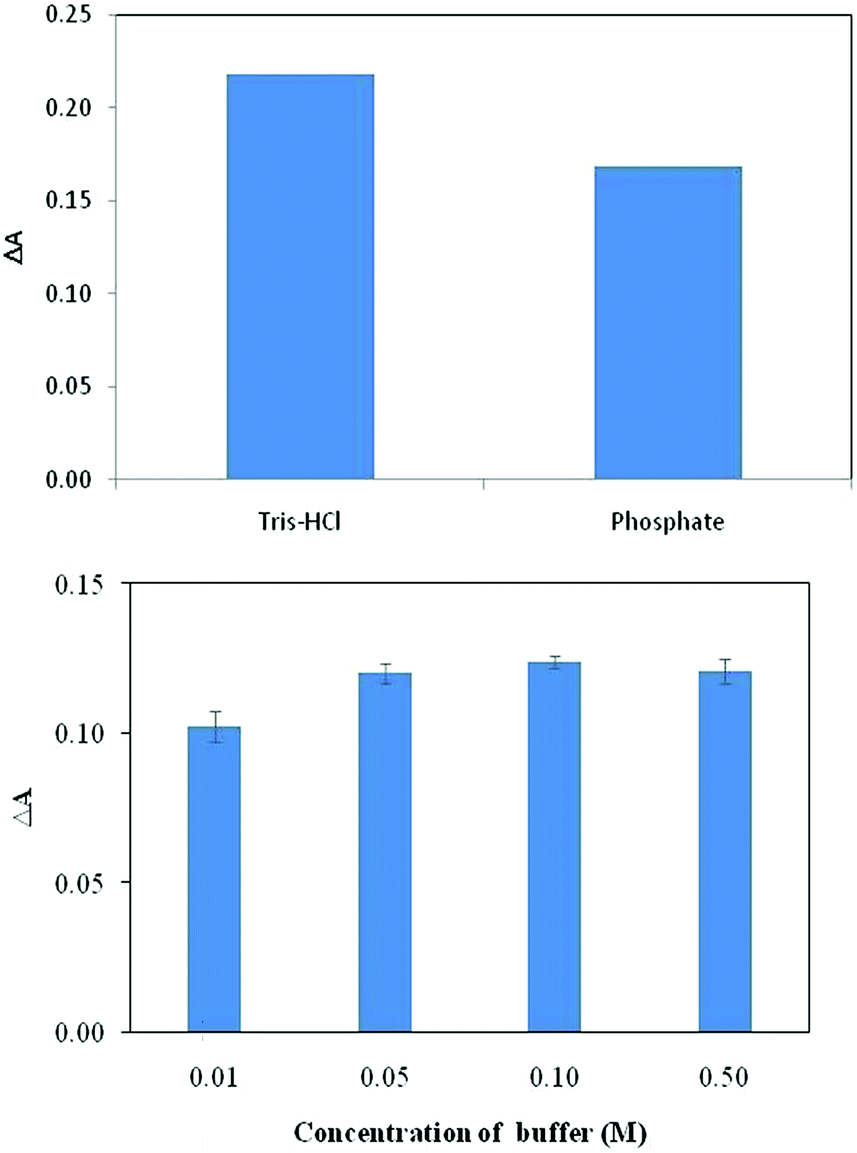 | ||
| Fig. 5 Effect of type of buffer (top) and concentration of buffer (bottom) on the response of the indicator to BSA for the dye concentration of 3.4 × 10−5 M. | ||
Spectrophotometric determination of BSA
As it was shown in (Fig. 2), addition of BSA to dye induced a decrease in the absorption of the dye at 541 nm in accompany with an increase in absorbance at 650 nm. We used the change in absorbance at 541 nm after addition of BSA (ΔA) as an analytical signal. Taking difference in absorbance cancelled the effect of changing in absorbance of dye resulted from small changes in dye's concentration through deriving the calibration curve and analysis of unknown samples.At optimum conditions, the calibration curve was obtained by monitoring the changes in absorbance of the dye as function of added concentration of BSA. The resulting calibration graphs of ΔA vs. [BSA] at pH 7.4 and pH = 9.3 are shown in (Fig. 6). As can be seen, similar linear behaviours were obtained in both pH media. Under optimized conditions, the calibration graphs were linear over two concentration ranges of 22.00–112.00 nM (1.46–7.54 mg L−1) and 0.43–4.10 μM (28.60–272.65 mg L−1) at pH = 7.4 and 10.8–53.5 nM (0.72–3.56 mg L−1) and 0.25–1.4 μM (16.6–93.0 mg L−1) at pH = 9.3.
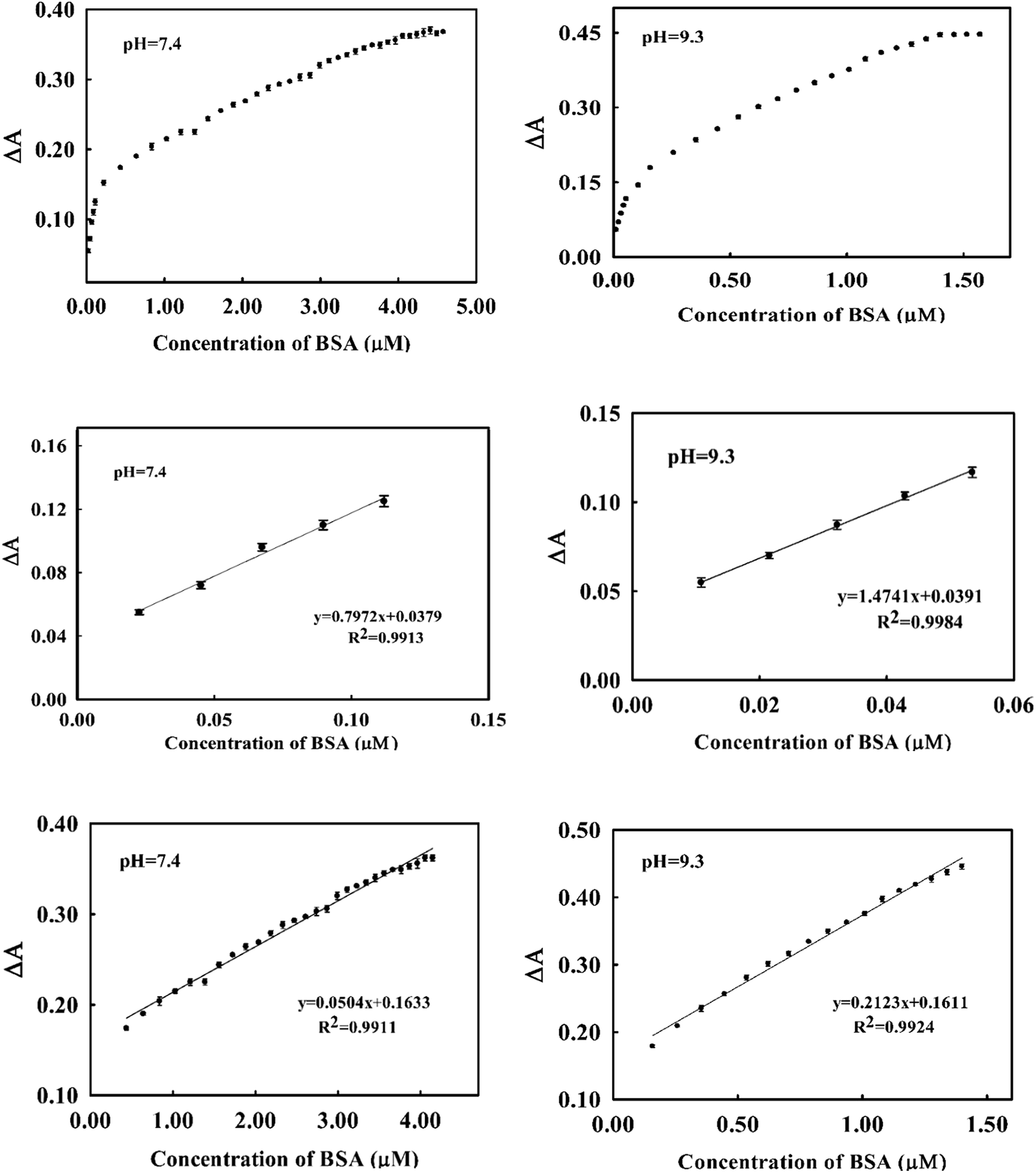 | ||
| Fig. 6 Calibration curves for the determination of BSA by spectrophotometric method at pH = 7.4 (left) and pH = 9.3 (right). In the top are shown the plot of whole concentration ranges and in the middle and bottom are the calibration graphs at two different linear ranges. | ||
The analytical appraisals of the suggested spectrophotometric method are listed in (Table 1). As seen, the limit of detection (3σ/m) was evaluated in pH = 7.4 as 6.25 nM (0.4 mg L−1) and in pH = 9.26 as 2.95 nM (0.19 mg L−1). It is obvious from Fig. 6 and Table 2 that the obtained results in both pH media are very similar. However, in biological pH, the slope of the calibration curve is a few lower than in basic pH, presumably due to the lower electrostatic interaction between cationic cyanine dye and negatively charged BSA.
| Figure of merit | pH = 9.3 | pH = 7.4 |
|---|---|---|
| a Relative standard deviation for 0.80 μM of BSA (n = 3). | ||
| Linear ranges | 10.8–53.50 nM | 22.00–112.00 nM |
| 0.25–1.40 μM | 0.43–4.10 μM | |
| Limit of detection | 2.95 nM | 6.25 nM |
| Calibration sensitivity | 1.0 × 106 M | 8.0 × 105 M |
| Analytical sensitivity | 2.2 × 108 | 1.3 × 108 |
| Limit of quantitation | 9.80 nM | 20.90 nM |
| RSDa (%) | 2.16 | 2.33 |
Determination of BSA by imaging method
For each image, three data matrices composed of color values of R, G and B were obtained. Each matrix was converted to a scalar by averaging of color values over all pixels. To avoid edge effect, only 250 pixels around the center of the image (forming a rectangular shape) were used for averaging. Thus, for each image (or solution) a vector of length of size 3 containing the averaged R, G and B color value was provided.To obtain calibration curves, each color value was plotted versus BSA concentration separately. It was found that the variation in relative color intensity of G with respect to BSA concentration was more fitted compared to the relative color intensities of R and B. Therefore G value was chosen as an analytical signal. Like spectrophotometric analysis, calibration curves were derived in Tris–HCl buffer of pH = 7.4 and 9.3 (Fig. 7). Two different linear ranges were obtained in both pH media. In pH = 9.3 the linear ranges were 18.00–53.00 nM and 0.25–1.40 μM with a limit of detection of 0.45 nM whereas in biological pH (pH = 7.4) the linear ranges were obtained in the concentration ranges of 170.00–700.00 nM and 1.70–23.00 μM. These values are similar to those obtained by the spectrophotometric method.
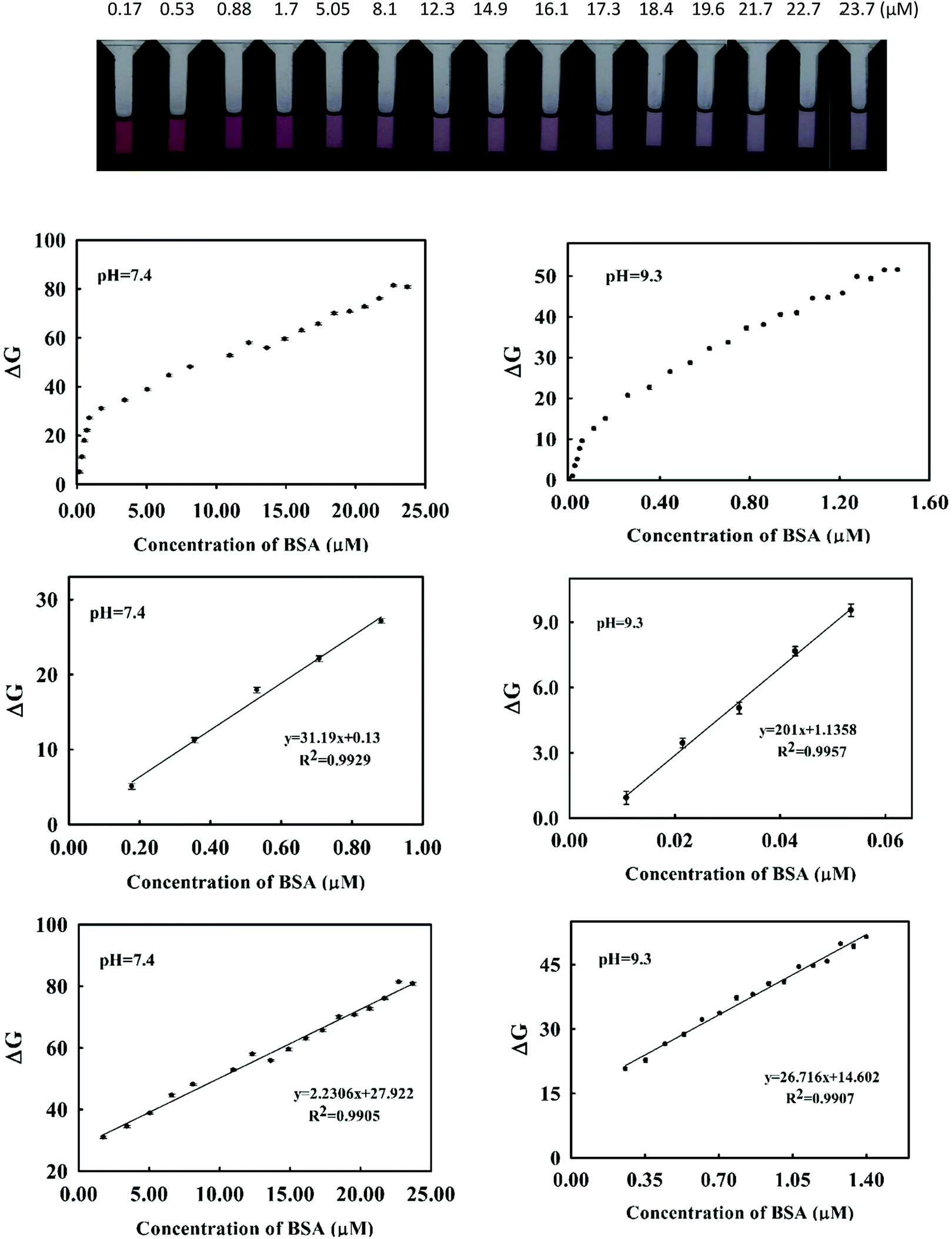 | ||
| Fig. 7 (Top) The photographs of the standard solutions of BSA of different concentrations in the presence of the cyanine dye. The plots are calibration curves for the determination of BSA by imaging method at pH = 7.4 (left) and pH = 9.3 (right). The top plots are for the whole concentration ranges and in the middle and bottom are the calibration graphs at two different linear ranges. | ||
To understand how the dye–BSA system changed along with time, the dye absorbance signal with time was explored. As shows in (Fig. 8), the dye absorbance signal changes slowly as time passed. Next, absorbance signal of the dye–BSA complex (at different concentrations of BSA) is highly stable for about 30 min. In this time period, the apparent BSA concentration changed less than 5%. Thus, in comparison to the previously reported methods14,15 the proposed method provides a larger range of time for accurate measurement of BSA concentration.
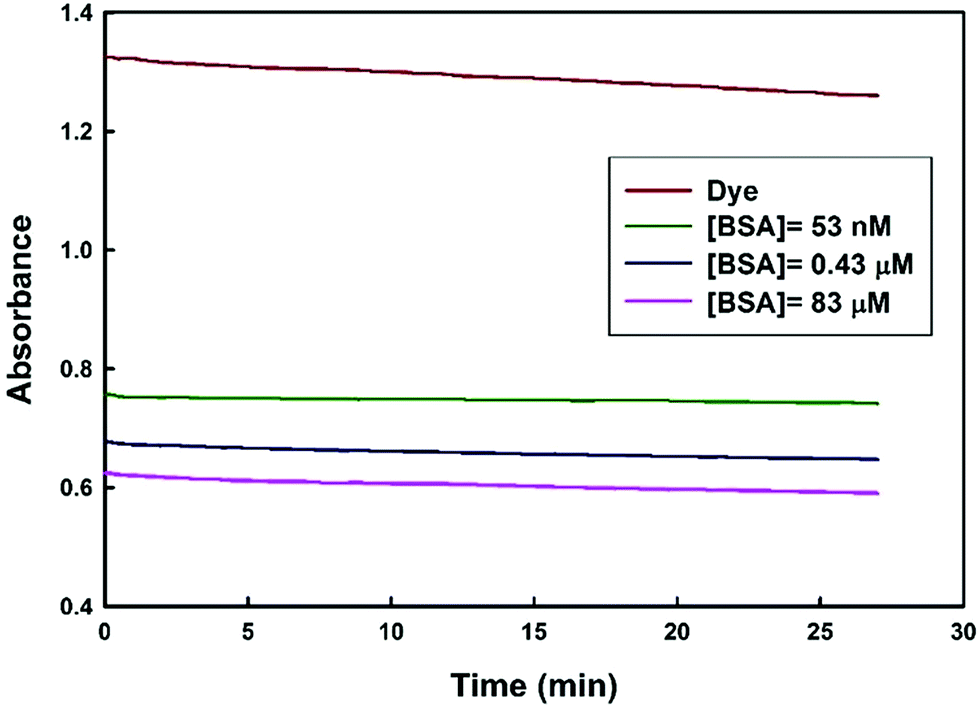 | ||
| Fig. 8 Changes in the absorbance of dye and dye–BSA complex at 541 nm as function of time. | ||
Total protein analysis
Most of the protein assay methods are non-selective and they response to total protein in the sample instead of individual protein. So, if the unknown protein sample has similar characteristics to the protein measured in the standard curve, the concentration of the unknown could be determined with a high degree of accuracy. In this work, to investigate the selectivity of the suggested method toward proteins, the response of the method was investigated for another protein (i.e., human serum albumin; HSA) and two protein mixtures (soluble lens protein and casein) in the same concentrations as BSA. The results are given in (Table 2). It is observed that, for two different individual proteins and two different protein mixtures, the proposed method resulted in the same response. This confirms that the method responds to total protein concentration and it is a non-selective protein assay method.Effect of foreign substances
For any given protein assay, however, further discrepancies may arise from non-protein interferences that produce protein overestimation, underestimation and a reduction of the linear response range. These sources of interference are best explained from the knowledge of the ionic dye species involved that most likely due to competition with the dye for protein and generate a colored complex themselves. Pervious method such as standard Lowry procedure is subject to interference by compounds like potassium ion, magnesium ion, urea, and carbohydrates, the relatively insensitive biuret reaction is subject to interference by Tris, ammonia and glucose.3Here, the selectivity of the method was evaluated by testing response of the dye to other compounds. To do so, changes in the absorbance spectra of dye–BSA mixture after addition of the interfering species were monitored. The addition of different concentrations of sodium, magnesium, calcium, chloride, oxalate, citrate, urea, creatinine, glucose and ascorbic acid to BSA were studied. It was observed that all species excluding ascorbic acid did not interfere in 1000 fold excess of BSA concentration. For ascorbic acid the tolerance limit was 500 fold excess of BSA concentration. As is seen in (Fig. 9), at concentration ratio of 1000/1 of the studied species, no significant changes were observed in the absorbance of dye–BSA system, explaining the high selectivity of the proposed method for proteins over other coexisting species.
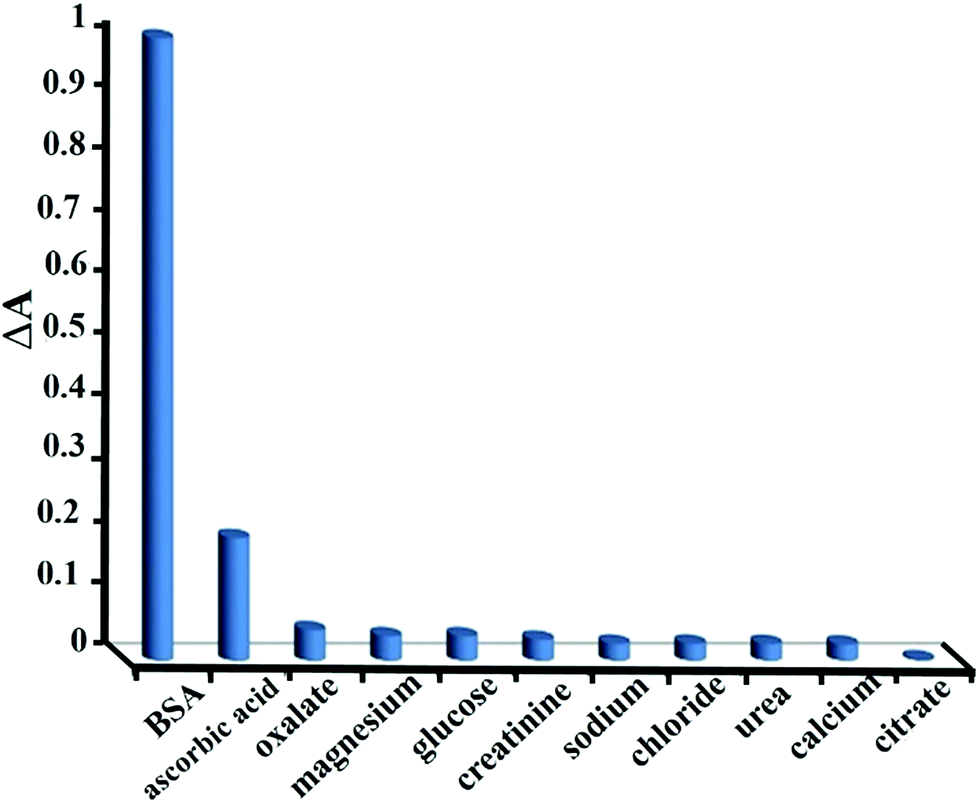 | ||
| Fig. 9 Selectivity of the suggested method for BSA over different species. | ||
Analysis of real samples
The suggested method was applied for assay of protein in artificial urine and blood plasma samples. There is a connection between the level of human serum albumin (HSA) in urine, blood plasma and some diseases, such as nephropathy, chronic liver diseases16 and unstable angina.17 The very high sensitivity of our method allows the analysis of low level amounts of protein in urine. Therefore, in addition to real blood plasma samples, the suggested method was employed for assay of protein in urine. To do so, artificial urine18 samples, spiked with trace amounts of HSA, were analyzed by the suggested method. A 1.0 mL portion of the sample solutions was analyzed using the standard addition method and the recoveries were evaluated (Table 3). As seen the method offers high level of accuracy with very low standard deviation for analysis of trace amounts of protein in urine.For the assay of protein in blood plasma by this new method, the samples were diluted appropriately to be within the linear range and then added to the buffered solution of the cyanine dye as was described in the Procedure section. To evaluate the accuracy of the method in analysis of real samples, the standard protein assay method based on bromocresol green reagent was adapted.19,20 Determination of the protein level in blood samples by both standard and new methods was achieved by standard addition. As it is shown in Table 4, there is an excellent agreement between the results of the new and standard methods.
Conclusion
In conclusion, to overcome the long-standing problem associated with the common protein assays for total protein, we developed a simple colorimetric method by spectroscopy and image analysis. A synthesized cationic cyanine dye represented a visual color change in the presence of BSA, enabling the detection of BSA in the concentration range of 22.00–112.00 nM and 0.43–4.10 μM in pH = 7.4 with detection limit of 20.9 nM. The proposed method is accurate, reproducible, and time-insensitive that provides a high-throughput means to measure HSA and BSA. This new method is more sensitive than the other methods and this superior sensitivity may be exploited in clinical medicine and biochemistry. In addition to great sensitivity, the method represented high accuracy in analysis of blood plasma as real samples.Acknowledgements
Financial support of this project by Shiraz University research council is highly appreciated.References
- J. R. Hoffman and M. J. Falvo, J Sports Sci. Med., 2004, 3, 118 Search PubMed.
- F. Kump and D. S. Booth, Protein Assays Handbook, 2008, p. 3 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265 CAS.
- K. P. Shrivastaw, S. Singh, S. B. Sharma and J. Sokhey, Biologicals, 1955, 23, 299 CrossRef PubMed.
- L. Lu, H.-Z. He, H.-J. Zhong, L.-J. Liu, D. S.-H. Chan, C.-H. Leung and D.-L. Ma, Sens. Actuators, B, 2014, 201, 177 CrossRef CAS.
- F. Shakerizadeh-shirazi, B. Hemmateenejad and A. M. Mehranpour, Anal. Methods, 2013, 5, 891 RSC.
- B. Hemmateenejad, F. Shakerizadeh-shirazi, S. Heidari and A. Shahrivar-kevishahi, Anal. Methods, 2015, 7, 6318 RSC.
- B. Hemmateenejad, N. Mobaraki, F. Shakerizadeh-shirazi and R. Miri, Analyst, 2010, 135, 1747 RSC.
- B. Hemmateenejad, S. Dorostkar, F. Shakerizadeh-shirazi and M. Shamsipur, Analyst, 2013, 138, 4830 RSC.
- E. L. Gelamo and M. Tabak, Spectrochim. Acta, Part A, 2000, 56, 2255 CrossRef.
- F. Samari, Ph.D thesis, Shiraz University, Shiraz, 2008.
- J. C. Miller and J. N. Miller, Statistic and Chemometrics for Analytical Chemistry, Multivariate analysis, 2005, ch. 8, p. 213 Search PubMed.
- S. E. Smith, J. M. Williams, S. Ando and K. Koide, Anal. Chem., 2014, 86, 2332 CrossRef CAS PubMed.
- J. E. Gustafsson, Clin. Chem., 1976, 22, 616 CAS.
- F. Wong, Nat. Rev. Gastroenterol. Hepatol., 2007, 4, 43 CAS.
- S. Ju, J. Ni, J. Su, M. Pan and J. Zhu, Lab. Med., 2008, 39, 668 CrossRef.
- S. Chutipongtanate and V. Thongboonkerd, Anal. Biochem., 2010, 402, 110 CrossRef CAS PubMed.
- A. M. Ramezani, J. Manzoori, M. Amjadi and A. Jouyban, Sci. World J., 2012, 2012, 1 CrossRef PubMed.
- B. T. Doumas, W. A. Watson and H. G. Biggs, Clin. Chim. Acta, 1971, 31, 87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25234b |
| This journal is © The Royal Society of Chemistry 2016 |