
Electrically stimulated liquid phase microextraction combined with differential pulse voltammetry: a new and efficient design for in situ determination of clozapine from complicated matrices†
Ahmad Rouhollahi,
Masoomeh Kouchaki and
Shahram Seidi*
Department of Analytical Chemistry, Faculty of Chemistry, K. N. Toosi University of Technology, P.O. Box 16315-1615, Tehran, Iran. E-mail: s.seidi@kntu.ac.ir; Fax: +98 21 88035187; Tel: +98 21 23064228
First published on 25th January 2016
Abstract
In this work, for the first time, a new, simple, low-cost and efficient design was presented for in situ determination of clozapine (CLZ) from human plasma samples by combination of electromembrane extraction (EME) with differential pulse voltammetry (DPV). The charged target analyte was extracted by applying a 200 V d.c. electrical potential from an acidic sample solution, through the supported liquid membrane (SLM) into an acidic acceptor solution, which was located in the lumen of a porous hollow fiber. 2-Nitrophenyl octyl ether (NPOE) was impregnated into the pores of the hollow fiber and used as the SLM. Three microelectrodes, an Ag/AgCl, a platinum wire, and a graphite pencil lead as the reference, counter and working electrodes, respectively, were fixed by passing through a septum and located into a pipette tip connected to the upper end of the hollow fiber for electrochemical analysis of CLZ. Central composite design and response surface methodology were used to optimize the main parameters influencing the extraction efficiency and analytical response (current) including pH of donor and acceptor phases, extraction time and electrical potential difference. Under the optimized conditions, the proposed method showed a wide linear range of 3–1500 ng mL−1 with a determination coefficient higher than 0.993. Extraction recovery of 42% was achieved which corresponded to a preconcentration factor of 114. Limits of detection and quantification of 0.9 ng mL−1 and 3.0 ng mL−1 were obtained, respectively. The intra- and inter-day precisions (RSD%, n = 3) were less than 3.5% and 6.7%, respectively. Finally, the proposed method was successfully applied for determination of CLZ from some human plasma samples.
1. Introduction
Schizophrenia is one of the chronic debilitating and complex neuropsychiatric disorders.1 In the last decades, the population with schizophrenia has grown and several drugs have been presented for the treatment of this disease to elevate the quality of patients' lives.2 In comparison with first-generation antipsychotics (FGAs), second-generation ones (SGAs) have been increasingly used for the treatment of patients with schizophrenia due to their better efficacy.3 Among SGAs, clozapine (CLZ) has an important role and has been successfully used for the treatment of refractory schizophrenia.2 Current pharmacologic guidelines suggest that the levels of CLZ in blood should be monitored for optimal dosage and low toxicity.4Determination of drugs in biological fluids is one of the main analytical challenges attributing to the low concentrations of drugs and complexity of their matrices. To achieve reliable data from bioanalysis, a robust sample preparation technique is vital. In all of the analytical processes, sample preparation has an important role to obtain accurate and sensitive results. Time consuming and utilization of large volumes of toxic and expensive organic solvents are the common disadvantages of the traditional sample pretreatment methods such as liquid–liquid extraction (LLE) and solid phase extraction (SPE). Therefore, during the last decades, a lot of attentions have been paid to the miniaturized sample preparation techniques.
Among different liquid phase microextraction techniques (LPME), hollow fiber liquid phase microextraction (HF-LPME) has been most frequently reported in literature.5 In HF-LPME, target analytes are extracted from an aqueous sample solution through a water immiscible organic solvent, impregnating into the pores of the hollow fiber which acts as the thin supported liquid membrane (SLM), into a microliter volume of an acceptor solution locating inside the lumen of hollow fiber. This technique suffers from the long extraction times, because of the analytes transfer in this technique is carried out by the passive diffusion.5 In order to overcome this limitation, electromembrane extraction (EME) was introduced in 2006 by Pedersen-Bjergaard et al.6
It is well known that charged chemical and biochemical substances migrate in solution under the application of an electrical field. This type of transport, which is called electrokinetic migration, is the basis for the electrophoresis. Frequently, isolation based on electrokinetic migration is carried out in an aqueous one-phase system such as electrodialysis. Alternatively, isolation based on electrokinetic migration may be accomplished in a two- or three-phase system such as EME. In the last case, an organic solvent is located between two aqueous phases and two electrodes are inserted in each phase. An electrical filed is imposed between two electrodes. Under applying the electrical filed, the charged ions migrate toward the electrodes with their opposite charge. In fact, the used instrumental set-up in EME is the same as HF-LPME, except two platinum electrodes which are inserted into both donor and acceptor phases and a d.c. electrical potential sustaining across the SLM. Moreover, against conventional three-phase HF-LPME, in three-phase EME, the pH of both donor and acceptor phases is adjusted acidic (for basic analytes) or alkaline (for acidic analytes) to convert the analytes to their ionic form and provides their migration ability under the electrical field.
In EME, the extraction time is considerably decreased in comparison with conventional HF-LPME because the relative distribution of the ion (KD) between the two phases can be altered by application of different voltages.7,8 When the analyte is reached to the interface of donor phase/SLM, it enters into the organic solvent as ionic form due to applying the electrical field and pass through SLM. The composition of organic solvent should be properly chosen to help the analyte retain in its ionic form during transport through SLM. It has been reported that protonated analytes have tendency to deprotonation and conversion to neutral form into a non-polar SLM.6 This tendency differs from an analyte to another one and reduces migration speed and extraction recovery of analytes through SLM under the electrical field.
Up to now, several analytical methods have been applied to monitor CLZ and its metabolites in plasma and serum including high performance liquid chromatography (HPLC) with various detection techniques such as UV,9 fluorimetric detection,10 amperometric detection11 and mass spectrometry,12 gas chromatography (GC) with mass spectrometry13 and spectrophotometry.14 However, spectrophotometric methods suffer from high interference and low sensitivity. On the other hand, chromatography methods require filtration, high cost equipment and reagents and also they are time consuming.15
Electroanalytical techniques are the alternatives which can be applied to overcome these drawbacks.16–18 In comparison with the above-mentioned techniques, electroanalytical methods have the advantages such as cost effective, easy application, high sensitivity and fast detection.16–18 Various electrochemical methods including cyclic voltammetry (CV) using a carbon nanotubes-sodium dodecyl sulfate (CNTs-SDS) modified carbon paste electrode (CPE),19 or a biosensor electrode, made of blended horseradish peroxidase cross-linked with glutaraldehyde and bovine serum in the matrix of a CPE,20 linear sweep voltammetry using multiwall carbon nanotubes (MWCNTs)/new coccine (NC) doped polypyrrole have been proposed for determination of CLZ.21 Recently, many scientists have attracted to the use of pencil lead graphite electrode (PGE) in different electroanalytical applications attributing to the advantages such as commercial availability, low cost and good rigidity.21,22
In this work, a new, simple and efficient design was developed by combination of EME and DPV and exploited for sensitive in situ determination of CLZ from human plasma samples. A PGE was used as the working electrode of a miniaturized three-electrode system locating into a channel of a micropipette tip. To the best of our knowledge, this is the first case in the peer-reviewed literature for combination of EME and DPV as a unique system for consecutive extraction and in situ determination of CLZ from biological fluids.
2. Experimental
2.1. Reagents and materials
Clozapine was purchased from Tehran Chimi Pharmaceutical Co. (Tehran, Iran). 2-Nitrophenyl octyl ether (NPOE), tris-(2-ethylhexyl) phosphate (TEHP) and di-(2-ethylhexyl) phosphate (DEHP) were provided from Fluka (Buchs, Switzerland). Dimethylformamide (DMF), nitrobenzene (NB), hydrochloric acid (37%, w/w) and potassium chloride were prepared from Merck (Darmstadt, Germany). All other reagents were of analytical grade and were used without further purification. Drug-free human plasma samples (blood group O+) were obtained from Iranian Blood Transfusion Organization (Tehran, Iran). Aqueous solutions were prepared with doubly distilled deionized water. All solutions were stored at 4 °C and protected from light.2.2. Preparation of microelectrodes
A platinum wire (o.d.: 0.5 mm) and a graphite pencil lead (o.d.: 0.5 mm, HB, Rotring, Germany) were used as the counter and working electrodes, respectively. A calibrated Ag/AgCl microelectrode was prepared according to literature and was used as the reference electrode.23 To this end, a fresh and hot agar gel including 1.0 mL of water, 0.30 g of KCl, and 0.030 g of agar was prepared. Once the agar was properly prepared, one end of a piece of polyethylene tube (length: 30 mm, o.d.: 1.4 mm and i.d.: 0.9 mm) was immediately dipped into the hot agar mixture. After 5 min, that the mixture was jellified at one end of the tube, it was filled by saturated KCl solution. Then, an Ag wire (i.d.: 0.5 mm), covered by a thin film of AgCl, was inserted into the KCl solution. Finally, this reference microelectrode was inserted into the saturated KCl solution for penetrating into the gel.The prepared microelectrode was calibrated before use as the reference electrode for DPV analysis. For this purpose, a 0.01 M solution of K4Fe(CN)6 was used as a compound that creates clear reversible redox peak and its cyclic voltammograms were recorded at the presence of the microelectrode and an Ag/AgCl/KCl(sat.) electrode as the references, respectively. In comparison with Ag/AgCl/KCl(sat.) electrode, the handmade reference microelectrode showed a little drift about 0.02 V to positive potential. Acceptable stability and reproducibility were observed for the calibrated reference microelectrode during the electrochemical analysis.
As shown in Fig. 1, the counter, working and reference electrodes were set close together and fixed by passing the electrodes through a vial septum. Lengths of 10 mm of the platinum and graphite electrodes were in contact with the solution. Three microelectrodes were washed with deionized water before each measurement.
 | ||
| Fig. 1 Schematic illustrations of the equipment used for extraction (A) and for in situ determination of CLZ by EME-DPV (B). | ||
2.3. Apparatus
All electrochemical experiments were performed using a computer controlled μAutolab potentiostat/galvanostat type III with a general-purpose electrochemical software operating system GPES version 4.9 (Ecochemie, Utrecht, Holland). Voltammetric measurements were carried out by the three electrodes described in the Section 2.2.A schematic presentation of the equipment used for EME procedure is shown in Fig. 1A. A glass vial with a height of 6.5 cm and an internal diameter of 3 cm was used. The porous hollow fiber applied for housing the acceptor solution and immobilizing the supported liquid membrane (SLM) was a PP Q3/2 polypropylene hollow fiber (Membrana, Wuppertal, Germany) with an internal diameter of 1200 μm, wall thickness of 200 μm and 0.2 μm pores. A 10 cm piece of hollow fiber was cut out and cleaned in acetone prior to use. Two platinum wires with the diameters of 0.2 and 0.5 mm were used as the electrodes and inserted into the acceptor phase and the sample solution, respectively. The electrodes were connected to a power supply model 8760T3 with a programmable output voltage in the range of 0–600 V and providing currents in the range of 0–500 mA from Paya Pajoohesh Pars (Tehran, Iran). Stirring the solution was performed by a Heidolph MR 3001K magnetic stirrer (Schwa, Germany).
2.4. Standard and real sample solutions
A stock solution containing 1.0 mg mL−1 of CLZ was prepared by dissolving of appropriate amount of the drug in 0.1 M HCl. Working standard solutions were prepared freshly by diluting of the stock solution to the required concentrations. The plasma samples were spiked with the convenient amounts of CLZ stock solution, diluted with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 using doubly distilled deionized water and their pH values were adjusted to 4.5 by dropwise addition of 0.2 M HCl solution. These sample solutions were directly exposed to EME, without extra treatment.
:6 using doubly distilled deionized water and their pH values were adjusted to 4.5 by dropwise addition of 0.2 M HCl solution. These sample solutions were directly exposed to EME, without extra treatment.
2.5. Extraction procedure
A 30 mL sample solution containing target analyte in 0.1 mM HCl was transferred into the sample vial. A 10 cm piece of the polypropylene hollow fiber was dipped into the organic solvent for 10 s to impregnate the organic liquid membrane in the pores of the hollow fiber wall. The excess of the organic solvent was gently wiped away using a piece of tissue-paper. Two micropipette tips, which were inserted through the rubber cap of the glass vial, were used as the holders for the hollow fiber, guiding tube for one of the platinum electrode (cathode) and electrochemical cell for determination of CLZ (Fig. 1). One end of the hollow fiber was connected to one of the micropipette tips, the acceptor solution (pH 4.5) was introduced into the lumen of the fiber by a medical syringe and then the other end of the hollow fiber was connected to the second micropipette tip. The platinum electrode with the diameter of 0.2 mm (cathode) was directed into the lumen of the hollow fiber. The other electrode (anode) was led directly into the sample solution in the center of the U-shaped hollow fiber. The electrodes were subsequently coupled to the power supply. The whole sample compartment was placed on the stirrer and agitated at 1000 rpm during the experiments. The power supply was switched on and EME was performed for a predetermined time. Then, the power supply was switched off and the electrodes were taken out of the sample solution and the lumen of the hollow fiber. Subsequently, the extracted analyte into the acceptor phase was transferred to the channel of the micropipette tip by air blowing using a medical syringe (Fig. 1B). Finally, three microelectrodes were inserted into the channel of the micropipette tip for in situ voltammetric analysis.2.6. Voltammetric measurement
Against the conventional working electrodes, in this work, pencil graphite electrode was directly used for electrochemical measurements with no polish or sonication in an ultrasonic bath. Firstly, the electrochemical behavior of CLZ was investigated by recording cyclic voltammograms in 0.1 mM HCl. The cyclic voltammograms were obtained at a scan rate of 40 mV s−1 and the potential was scanned from +0.2 V to +0.8 V. Then, the differential pulse voltammetry was applied as the detection technique. Differential pulse voltammograms were recorded in the potential range of +0.2 V to +0.8 V, scan rate of 30 mV s−1 and pulse amplitude of 40 mV. The parameters for evaluation of the analytical performance, such as limit of detection (LOD), limit of quantification (LOQ), repeatability, calibration curve, etc., were investigated by utilizing differential pulse voltammograms.2.7. Data analysis
To achieve the optimum conditions for extraction of CLZ by EME, a central composite design (CCD) was employed. For this purpose STATISTICA software trial version 8.0 (StatSoft, Tulsa, OK, USA) was applied to generate the experimental matrix and evaluate the obtained results.3. Result and discussion
3.1. Voltammetric behavior of CLZ on PGE
The cyclic voltammograms of CLZ on a PGE is shown in Fig. 2. As can be seen, no obvious peak was observed for the blank solution (0.1 mM HCl) at PGE. In the case of CLZ (100 μM CLZ in 0.1 mM HCl), one pair of well-defined redox peaks appeared. The oxidation peak of CLZ located at +0.53 V vs. the calibrated reference microelectrode. According to the curve b in Fig. 2, it can be concluded that the extent and sensitive oxidation peak of CLZ is favorable for detection. | ||
| Fig. 2 Cyclic voltammograms of (the green line) 0 M CLZ, (the blue line) 100 μM CLZ in 0.1 mM HCl on PGE with scan rate of 40 mV s−1. | ||
3.2. Optimization of pulse amplitude and scan rate of DPV for electrooxidation of CLZ
As well known, the pulse amplitude does not affect the scan rate. However, large pulse amplitudes lead to an increase of the peak current signal. In other words, determination sensitivity is affected by the pulse amplitude which can be attributed to the more difference between the peak currents before and after pulse performing. On the other hand, the peak currents can be mutilated at very large amplitudes due to the non-linearity effects.24 Therefore, pulse amplitudes of 20 mV, 30 mV, 40 mV, 60 mV and 80 mV were selected to investigate the effect of this parameter on the peak current signal. The best result was obtained at the pulse amplitude of 40 mV and after that the peak current remained constant (Fig. 1S†). Therefore, the pulse amplitude of 40 mV was selected for further experiments.According to literature, scan rate is an important parameter which should be considered in DPV analysis because the current peak is directly proportional to the rate of electrolysis at the electrode surface.24 On the other hand, high scan rates lead to decreasing of analytical measurements precision due to appearance of asymmetric oxidation peaks.24 The scan rate is calculated by dividing of the potential step to the interval time which both of them are device parameters. The practical range of the scan rate in DPV is very narrow in comparison with CV. For example, a scan rate of 30 mV s−1 can be achieved by applying a potential step of 6 mV and interval time of 0.2 s. A series of scan rates including 5 mV s−1, 10 mV s−1, 15 mV s−1, 20 mV s−1, 30 mV s−1 and 40 mV s−1 were investigated. The results showed that the scan rate of 30 mV s−1 was the best choice by considering the oxidation peak current and the peak resolution (Fig. 2S†). Consequently, 30 mV s−1 was selected as the optimum value for subsequent experiments.
3.3. Optimization of electromembrane extraction of CLZ
Different variables influence the extraction efficiency of EME, including membrane organic solvent, pH of donor and acceptor phases, extraction time, voltage, stirring rate and ionic strength. In this study, experimental design and response surface methodology (RSM) were applied to find the optimum values of the variables affecting the extraction efficiency of EME with the aim of reducing the extraction time and process cost. Separate study of membrane organic solvent not only gives the best SLM but also provides simplicity of the design and reduces run numbers of the experimental matrix. Therefore, this parameter was separately optimized at first. Furthermore, the influences of ionic strength and stirring rate of the sample solution were evaluated, separately.As well known, stirring rate plays an important role to promote the kinetics and efficiency of extraction in EME by reducing the thickness of the double layer around SLM and increasing the rate of mass transfer. According to literature, stirring rate is more effective for the large volumes of sample solution.25 Regarding 30 mL volume of the sample solution in this work, the maximum stirring rate of 1000 rpm was chosen as the best amount. Increasing of stirring speed higher than 1000 rpm was not experimentally possible due to formation of intense vortex and bubble formation into the sample solution.
To investigate the effect of ionic strength, sodium chloride was added into the donor phase at two concentration levels of 1.25% and 2.5% (w/v). The results showed a negative effect on the extraction efficiency of EME by increasing the ionic strength of the sample solution. According to the previous studies, increasing the ionic substances into the sample solution leads to increasing of the ion balance value (χ) which is defined as the ratio of the total ionic concentration in the sample solution to that in the acceptor solution.26 This increases the competition among interferences ions and target analytes for migration through the SLM toward the acceptor phase and consequently, decreases the analyte flux across the SLM.26 Moreover, increasing of χ may lead to instability of SLM during extraction procedure due to Joule heating phenomenon.25 Thus, the extraction efficiency of EME would be more effective in the absence of salt.
Finally, the influences of the other parameters (pHs of donor and acceptor phases, voltage and extraction time) were evaluated by an experimental design method using a rotatable central composite design (RCCCD). During all optimization process, the concentration of CLZ was considered 50 μg L−1.
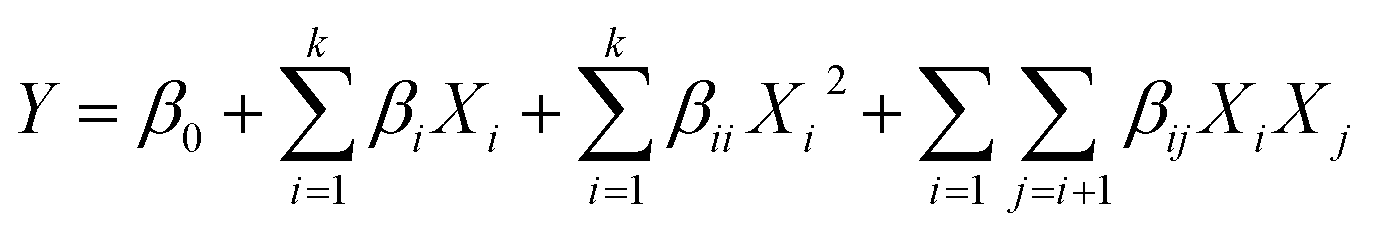 | (1) |
| N = 2f + 2f + Cp | (2) |
The peak current for each run was selected as the response objective for the study. The data obtained were evaluated by analysis of variance (ANOVA). A P-value less than 0.05 in the ANOVA table indicates the statistical significance of an effect at 95% confidence level. By using multiple regression analysis, the experimental responses were correlated with the four experimental factors. The model was described as follows:
| Current = 1.96933 + (0.00653 × A) + (0.22941 × B) + (1.66054 × C) − (3.48063 × D) − (0.000227 × AB) + (0.00367 × AC) − (0.000779 × AD) − (0.04596 × BC) + (0.032838 × BD) − (0.12856 × CD) − (0.0000310 × A2) − (0.00343 × B2) − (0.1162 × C2) + (0.39355 × D2) |
The ANOVA results for the quadratic regression model are shown in Table 1S.† As can be seen, the F-value of the model (19.48) is much greater than the tabular F-value of 2.51 (14, 12, 0.05) which implies the model is significant. Also, the F-value of 2.30 for the “lack of fit” indicates that it is not significant relative to the pure error. The coefficient of determination (R2) and adjusted R2 values were 0.9579 and 0.9087, respectively. This indicates that the model could explain 95.79% of the variability in the peak current response. Statistical significance was evaluated on the basis of the magnitudes of coefficients in the regression equation. As can be seen, the linear and quadratic terms of the acceptor phase pH (D), and donor phase pH (C) have the largest influences on the response. The next most significant factors were linear term of extraction time (B) and its interactions with pHs of donor phase (BC) and acceptor phase (BD) as well as interaction between voltage and acceptor phase pH (AD).
RSM was applied to analyze the effect of independent variables on the response. Fig. 3 illustrates the relationship between the explanatory and response variables in a three-dimensional representation of the response surface. To this end, two variables were varied within the experimental range and the others were kept at their central levels. The observed curvatures in RSM plots indicate interactions among experimental variables. Fig. 3 also depicts two-dimensional contour plot on the basis of the model equations which display the interaction between independent variables and assist in determining the optimum operating condition for the desirable responses. Based on the analysis and presented plots in Fig. 3, it can be observed that the response is increased by increasing the voltage and extraction time (Fig. 3A) to determined levels and declined thereafter. According to literature,29 in EME, the flux of the analytes across SLM into the acceptor solution is greatly dependent upon the applied voltage. Application of high electrical potentials not only increases the mass transfer rate of analytes through the artificial liquid membrane but also reduces deprotonation opportunity of the protonated analytes during transportation via SLM as a non-polar medium.30 Time is another parameter which can affect the flux of analytes in EME. The extraction recovery is increased by increasing both of time and voltage however; there are an antagonistic effect between time and voltage so that an increasing in the extraction time limits the increasing of voltage and vice versa. This fact can be attributed to increasing the possibility of electrolysis reactions into both donor and acceptor solutions which the last one may be resulted to back-extraction of analytes from acceptor phase into SLM, increasing the probability of Joule heating and consequently instability of SLM as well as decreasing the repeatability of extraction. Therefore, simultaneous investigation of time and voltage in EME provides this opportunity to find the best optimum values for these parameters. According to the RSM plots and statistical data, voltage of 220 V and extraction time of 18 min were selected as the optimum values of these variables.
 | ||
| Fig. 3 Response surfaces and contour plots of CLZ using RCCD which illustrate the relationships between the independent variables and the experimental response. In each case, two variables were varied within the experimental range and the others were kept at their central levels. | ||
In EME, pH of the donor phase should be adjusted at a value to convert the analytes in their ionized form providing the possibility of electrokinetic migration by applying an electrical driving force. Thus, for basic drugs such as CLZ, sample solution should be acidified to form analytes with positive charges. However, an important point which should be considered in the case of basic analytes is that increasing of the concentration of proton ions in donor phase leads to competition among proton and charged analytes for migration toward the negative electrode (cathode) located into the acceptor phase. Therefore, extraction recovery may be decreased in comparison with sample solutions containing less proton ions.31 Increasing of proton ions concentration in acceptor phase increases the rate of analytes releasing in the acceptor phase/SLM interface and so extraction efficiency. However, there are some limitations for this issue.
Increasing content of ions in each of donor and acceptor phase leads to increasing the numbers of ions migrate through SLM at a given moment, increasing of Joule heating and instability of SLM and consequently increasing electrolysis reactions on the surfaces of electrodes. Electrolysis reactions lead to bubble formation into both donor and acceptor phases and increasing uncertainties in the obtained data by EME. Another drawback which is followed by increasing the concentration of other ions such as protons into sample solution and acceptor phase is increasing the thickness of double layer around SLM, increasing the analytes mass transfer resistance and thus decreasing of extraction efficiency.
As can be seen in RSM and contour plots, the best response was obtained at the donor phase pH of 4.5 and acceptor pH of 2.0. These values can be explained by considering the pKa values of CLZ32 3.70 and 7.60, as well as the mentioned points above. At pH of 4.5, CLZ exists as monocharge whereas at pHs lower than 3.7 it contains two positive charges which decrease the extraction efficiency. Although, the migration ability under an electrical field is increased by increasing the charge density but also mass transfer resistance through SLM as a non-polar medium and consequently the thicknesses of double layer around SLM is increased. On the other hand, the optimum pH of 2.0 for the acceptor phase can be explained by converting of CLZ to its double charges form at the SLM/acceptor phase interface and so increasing the releasing rate. Therefore, pH values of 4.5 and 2.0 were chosen as the optimum values for the subsequent experiments.
3.4. Analytical performance
The optimized conditions were performed to study the applicability of the proposed method for extraction and determination of CLZ. LOD, LOQ, linearity, preconcentration factor (PF), extraction recovery (ER%) and precision of EME-DPV were assessed for water solutions spiked with CLZ at various concentrations. LOD value was obtained based on practical experiments. For this purpose, the concentration of analyte was reduced since the obtained EME-DPV response for CLZ created detectable signals-to-noise ratio of 3 (3S/N). LOQ value was considered the low concentrations that linearity of the calibration curve was started from it. The voltammograms and calibration curves were recorded and plotted as shown in Fig. 4. The DPV peak currents against the concentrations of CLZ were distinguished in two linearity ranges. As can be seen, the slop ratios of the calibration curves in two linearity ranges have a considerable difference. This fact has been reported in literature and can be attributed to the saturation of the working electrode surface at high concentration of the electroactive analyte.33 The analytical characteristics are summarized in Table 1. CLZ demonstrated wide linearity range with correlation coefficient values greater than 0.993. PF was calculated as the ratio between the final analyte concentration in the acceptor phase and the initial concentration of analyte in the sample solution. ER% is defined as the percentage of the moles number of analyte extracted into the acceptor phase to that originally present in the sample solution. Repeatability (intra-day) precision was assessed by extracting three replicates of CLZ at the concentration level of 50 μg L−1 and reproducibility (inter-day) precision was evaluated by extracting the same sample over three consecutive days. | ||
| Fig. 4 DPV voltammograms of various CLZ concentrations (down to up: 3, 6, 8, 10, 25, 50, 100, 200, 500, 1000 and 1500 ng mL−1) in the optimal conditions, the dependences between peak current (μA) and CLZ concentrations (ng mL−1) appear in the inset. | ||
| Evaluation parametera | Value | |
|---|---|---|
| a Enrichment factor, extraction recovery and RSD were calculated based on extraction of 50 ng mL−1 of CLZ (n = 3). | ||
| Limit of detection (LOD) | 0.9 ng mL−1 | |
| Limit of quantification (LOQ) | 3.0 ng mL−1 | |
| Dynamic linear range (DLR) | Linear range 1 | 3–10 ng mL−1 |
| Linear range 2 | 10–1500 ng mL−1 | |
| Determination coefficient (R2) | >0.993 | |
| Preconcentration factor (PF) | 114 | |
| Extraction recovery% (ER%) | 42 | |
| RSD% (n = 3) at the concentration of 50 ng mL−1 | Intra-day | 3.5 |
| Inter-day | 6.7 | |
The selectivity in EME can be provided by several factors including the applied voltage, type of organic solvent used as SLM, extraction time and pH of the sample solution. For basic analytes, acidic compounds are not extracted into the acceptor solution, neutral compounds are efficiently discriminated because they do not migrate under the electrical field, and only basic compounds can enter into SLM. Moreover, the relative distribution of the ions (KD) at the sample solution/SLM interface and consequently, the extraction selectivity can be altered by application of different voltages.8,29 Also, it has been found that composition of the organic solvent effectively controls the extraction selectivity.29,34,35 Time is another parameter which can affect selectivity because mass transfer across SLM is the rate limiting step in EME.36 pH of the sample solution can also affect the charge of analytes and their electrokinetic migration under the electrical field. In addition, the range of potential that the oxidation of the target analyte is occurred during DPV is different from the ranges for other compounds and consequently, combination of DPV with EME helps to the improvement of determination selectivity. Finally, an analyte-free plasma sample was extracted by EME. Determination of the extract by DPV did not show any obvious peak.
The analytical parameters of the PGE electrode was compared with other modified electrodes reported in literature for determination of CLZ (Table 2S†). PGE, along with its simplicity, availability and low cost, presented better or comparable LOD and linearity ranges.
Also, comparison of EME-DPV with other extraction techniques, applied for determination of CLZ, is provided in Table 2. In comparison with other methods, EME-DPV not only has lower consumption of organic solvent and cost equipment as well as easier operation but also provided better or comparable LOD and linearity range.
| Method | Linearitya | LODa | Volume of organic solvent (μL) | Ref. |
|---|---|---|---|---|
| a All concentration are based ng mL−1.b Microextraction packed sorbent-high performance liquid chromatography.c Solid phase extraction-gas chromatography-mass spectrometry.d Liquid–liquid extraction-capillary electrophoreses-UV detection.e Liquid–liquid extraction-ultra-high performance liquid chromatography-tandem mass spectrometry.f Solid phase extraction-liquid chromatography-mass spectrometry.g Solid phase extraction-high performance liquid chromatography-UV detection.h Electromembrane extraction-differential pulse voltammetry. | ||||
| MEPS-HPLC-Coulometricb | 2.5–1000 | 0.08 | 150 | 37 |
| SPE-GC-MSc | 3–600 | 0.45 | 2000 | 13 |
| LLE-CE-UVd | 50–800 | 5 | 500 | 38 |
| LLE-UPLC-MS/MSe | 50–1500 | 0.5 | — | 39 |
| SPE-LC-MSf | 10–1000 | 0.3 | 10000 |
40 |
| SPE-HPLC-UVg | 20–2500 | 7 | 1000 | 9 |
| EME-DPVh | 3–1500 | 0.9 | 90 | This work |
3.5. Real sample analysis
Major problem in the analysis of biofluids is electrochemical reactivity of biological species (such as ascorbic acid and uric acid) which cause increasing the background signal and fouling of the electrode surface.41 EME technique has excellent capability to eliminate the interferences because with locating of the negative electrode into the acceptor phase only positive charged compounds can migrate through SLM into the acceptor phase, hence, the negative and neutral ones remain in the sample solution. According to the obtained conditions for EME-DPV, in the donor phase (pH = 4.5) ascorbic acid is converted in its molecular form and could not migrate under electrical field whereas it may directly interact with CLZ and decrease its analytical signal.41,42 In addition, NPOE which was used as SLM, leads to extraction of only nonpolar compounds with logP values greater than one. Also, uric acid (logP = −1.107) does not have tendency toward NPOE and thus could not be extracted.
The ability of the developed method to detect CLZ in human plasma with minimal sample preparation was examined. Three different plasma samples were spiked at different concentrations of CLZ including 50, 300 and 700 μg L−1. These concentrations are close to the upper and lower optimal therapeutic range (350–1000 μg L−1) of CLZ. The results are summarized in Table 3. Matrix effect%, accuracy and RSD% were calculated for the spiked samples.
| Sample | Cinitiala | Caddedc | Cfoundc | Matrix effect% | Error% | RSDd% |
|---|---|---|---|---|---|---|
| a Match matrix was used to determine initial concentration in plasma samples.b N.D: not detected.c All concentration are based on ng mL−1.d RSD% values were calculated for n = 3. | ||||||
| Plasma 1 | N.Db | 50 | 16.5 | 33 | −5 | 6.1 |
| Plasma 2 | N.D | 300 | 93 | 31 | −3 | 6.4 |
| Plasma 3 | N.D | 700 | 238 | 34 | −9 | 5.8 |
By definition of Food and Drug Administration (FDA), a matrix effect is the direct or indirect alteration or interference in response due to the presence of unintended analytes or other interfering substances in the sample. The matrix effect was calculated by comparing the analytical current in spiked water (neat solution) with those that obtained for the spiked plasma samples. The results in the range of 31–34% indicated the existence of matrix effect for the plasma samples and thus a match matrix method was used for the quantitative determinations.
To examine the accuracy of the proposed method, relative recovery (RR%) values were calculated by comparing the analytical currents obtained at the concentrations of 50, 300 and 700 μg L−1 in plasma 1, 2 and 3, respectively with the corresponding currents resulted in a drug-free plasma sample which used as the match matrix for the quantitative determinations (eqn (3)).
 | (3) |
The spiked recoveries were obtained in the range of 91–97%. Also, the calculated RSD% values for determination of CLZ in the plasma samples were less than 6.4%. These results demonstrated a good performance and accuracy of EME-DPV for determination of CLZ in plasma samples.
4. Conclusion
In the present work, for the first time, EME in combination with DPV was developed as a new, simple, cost effective and unique design for extraction and in situ determination of CLZ from plasma sample. Different parameters affecting the extraction efficiency of EME were optimized by applying central composite design. A PGE was used as the working electrode of a three-microelectrode system, locating into a channel of a micropipette tip, for electrochemical detection of CLZ. The properties of PGE such as easy accessible and low cost are very useful in the function of inexpensive electrochemical devices for analysis of pharmaceutical compounds. Also, in situ determination provides more simplicity, sensitivity and repeatability as well as decreasing the analysis time. Moreover, this new design can be applied as a portable system for in field analysis. Overall, beside the low cost of EME-DPV, the obtained results indicated suitable sensitivity, wide linearity, good repeatability, high sample cleanup and easy operation for the proposed method.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
The authors gratefully acknowledge K. N. Toosi University of Technology (Tehran, Iran).References
- M. H. Mashhadizadeh and E. Afshar, Electrochim. Acta, 2013, 87, 816–823 CrossRef CAS.
- Z. Ismail, A. M. Wessels, H. Uchida, W. Ng, D. C. Mamo, T. K. Rajji, B. G. Pollock, B. H. Mulsant and R. R. Bies, Am. J. Geriatr. Psychiatr., 2012, 20, 53–60 CrossRef PubMed.
- M.-L. Lu, T.-N. Wang, T.-Y. Lin, W.-C. Shao, S.-H. Chang, J.-Y. Chou, Y.-F. Ho, Y.-T. Liao and V. C.-H. Chen, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2015, 58, 47–50 CrossRef CAS PubMed.
- H. Ben-Yoav, S. E. Chocron, T. E. Winkler, E. Kim, D. L. Kelly, G. F. Payne and R. Ghodssi, Electrochim. Acta, 2015, 163, 260–270 CrossRef CAS.
- S. Cui, G. Ouyang, G. Duan, J. Hou, T. Luan and X. Zhang, J. Chromatogr. A, 2012, 1266, 10–16 CrossRef CAS PubMed.
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2006, 1109, 183–190 CrossRef CAS PubMed.
- Y. Yamini, S. Seidi and M. Rezazadeh, Anal. Chim. Acta, 2014, 814, 1–22 CrossRef CAS PubMed.
- C. J. Collins and D. W. M. Arrigan, Anal. Bioanal. Chem., 2009, 393, 835–845 CrossRef CAS PubMed.
- L. Mercolini, F. Bugamelli, E. Kenndler, G. Boncompagni, L. Franchini and M. A. Raggi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 846, 273–280 CrossRef CAS PubMed.
- R. Waschgler, M. R. Hubmann, A. Conca, W. Moll and P. König, Int. J. Clin. Pharmacol. Ther., 2002, 40, 554–559 CrossRef CAS PubMed.
- M. A. Raggi, F. Bugamelli, C. Sabbioni, D. De Ronchi, S. Pinzauti and V. Volterra, Chromatographia, 2000, 51, 147–153 CAS.
- E. Choong, S. Rudaz, A. Kottelat, D. Guillarme, J.-L. Veuthey and C. B. Eap, J. Pharm. Biomed. Anal., 2009, 50, 1000–1008 CrossRef CAS PubMed.
- I. Vardakou, A. Dona, C. Pistos, G. Alevisopoulos, S. Athanaselis, C. Maravelias and C. Spiliopoulou, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2327–2332 CrossRef CAS PubMed.
- A. A. Mohamed and S. M. Al-Ghannam, Il Farmaco, 2004, 59, 907–911 CrossRef CAS PubMed.
- E. Dede, Ö. Sağlam and Y. Dilgin, Electrochim. Acta, 2014, 127, 20–26 CrossRef CAS.
- H. Ahmar, A. R. Fakhari, H. Tabani and A. Shahsavani, Electrochim. Acta, 2013, 96, 117–123 CrossRef CAS.
- A. R. Fakhari, A. Sahragard, H. Ahmar and H. Tabani, J. Electroanal. Chem., 2015, 747, 12–19 CrossRef CAS.
- H. Ahmar, H. Tabani, M. H. Koruni, S. S. H. Davarani and A. R. Fakhari, Biosens. Bioelectron., 2014, 54, 189–194 CrossRef CAS PubMed.
- J. G. Manjunatha, B. E. K. Swamy, G. P. Mamatha, O. Gilbert, M. T. Srinivas and B. S. Sherigara, Der Pharma Chemica, 2011, 3, 236–249 CAS.
- B. Blankert, O. Dominguez, W. El Ayyas, J. Arcos and J. M. Kauffmann, Anal. Lett., 2005, 37, 903–913 CrossRef.
- S. Shahrokhian, Z. Kamalzadeh and A. Hamzehloei, Bioelectrochemistry, 2013, 90, 36–43 CrossRef CAS PubMed.
- J. I. Gowda and S. T. Nandibewoor, Electrochim. Acta, 2014, 116, 326–333 CrossRef CAS.
- M. Saraji, B. Farajmand, A. A. Ensafi, A. R. Allafchian and Z. M. Zare, Talanta, 2010, 82, 1588–1593 CrossRef CAS PubMed.
- C. R. T. Tarley and L. T. Kubota, Anal. Chim. Acta, 2005, 548, 11–19 CrossRef CAS.
- S. Seidi, Y. Yamini, M. Rezazadeh and A. Esrafili, J. Chromatogr. A, 2012, 1243, 6–13 CrossRef CAS PubMed.
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1174, 104–111 CrossRef CAS PubMed.
- M. Balchen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1152, 220–225 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini, T. Baheri and R. Feizbakhsh, J. Chromatogr. A, 2011, 1218, 3958–3965 CrossRef CAS PubMed.
- T. M. Middelthon-Bruer, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Sep. Sci., 2008, 31, 753–759 CrossRef CAS PubMed.
- A. Gjelstad, T. M. Andersen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1157, 38–45 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini, A. Heydari, M. Moradi, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS PubMed.
- K. Farhadi and A. Karimpour, Anal. Sci., 2007, 23, 479–483 CrossRef CAS PubMed.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Anal. Chim. Acta, 2008, 618, 54–60 CrossRef CAS PubMed.
- A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2006, 1124, 29–34 CrossRef CAS PubMed.
- S. Seidi, Y. Yamini and M. Rezazadeh, J. Pharm. Biomed. Anal., 2011, 56, 859–866 CrossRef CAS PubMed.
- A. Gjelstad, H. Jensen, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2012, 742, 10–16 CrossRef CAS PubMed.
- M. A. Saracino, G. Lazzara, B. Prugnoli and M. A. Raggi, J. Chromatogr. A, 2011, 1218, 2153–2159 CrossRef CAS PubMed.
- Y.-H. Ho, W.-K. Ko, H.-S. Kou, H.-L. Wu and S.-M. Wu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 809, 111–116 CrossRef CAS PubMed.
- L. Patteet, K. E. Maudens, Z. Vermeulen, G. Dockx, M. De Doncker, M. Morrens, B. Sabbe and H. Neels, Clin. Biochem., 2014, 47, 336–339 CrossRef CAS PubMed.
- H. A. G. Niederländer, E. H. M. Koster, M. J. Hilhorst, H. J. Metting, M. Eilders, B. Ooms and G. J. De Jong, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 834, 98–107 CrossRef PubMed.
- H. Ben-Yoav, T. E. Winkler, E. Kim, S. E. Chocron, D. L. Kelly, G. F. Payne and R. Ghodssi, Electrochim. Acta, 2014, 130, 497–503 CrossRef CAS.
- I. K. Kiplagat, T. K. O. Doan, P. Kubáň and P. Boček, Electrophoresis, 2011, 32, 3008–3015 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra25157e |
| This journal is © The Royal Society of Chemistry 2016 |