
Mechanistic insights into the ring-opening of biomass derived lactones†
Abstract
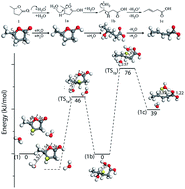
Deoxygenation of biomass-derived lactone molecules such as γ-valerolactone (GVL) by catalytic ring-opening and decarboxylation facilitate the production of a variety of fuels and chemicals. Density functional theory (DFT) calculations were performed to reveal the mechanism of the ring-opening step. In order to elucidate the effect of substituents and ring-size on the rate of the ring-opening step, lactone molecules such as γ-butyrolactone (GBL), γ-caprolactone (GCL), δ-valerolactone (DVL) and ε-caprolactone (ECL) were included. DFT calculations show that the ring-opening reaction proceeds via the formation of a stable oxocarbenium intermediate. Subsequently, the ring-opening occurs through a two-step mechanism to yield unsaturated acids. The intrinsic barriers for the two-step reaction in GVL were estimated to be 69 and 48 kJ mol−1 respectively which are comparable to the experimentally observed apparent activation energies. On changing the ring-size from 5 to 7 member ring lactones (GBL, DVL and ECL), the activation energy for the ring-opening steps was observed to follow the trend predicted by the strain theory. In contrast, on changing the substituent at C4 of the 5-member ring lactones (GBL, GVL and GCL), the activation energies for ring-opening are dictated by a combination of inductive and steric effects. It is expected that the stability of the oxocarbenium ion formed will have a significant role on the rate of the ring-opening. The estimated rates for the ring-opening step with respect to the ring-size show a direct correlation with the enthalpy of oxocarbenium ion formation in the gas phase. Further investigation by ab initio molecular dynamics simulations in implicit and explicit aqueous environment indeed show stable oxocarbenium ions formed which were observed to remain intact for greater than 2 ps of simulation time.
 Please wait while we load your content...
Please wait while we load your content...

