
A comparative study of the CO oxidation reaction over pristine and C-doped boron nitride fullerene†
Abstract
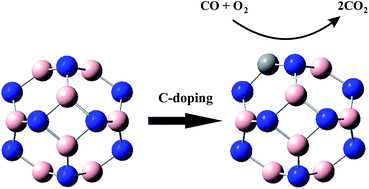
In this work, we employ density functional theory calculations to investigate the CO oxidation mechanisms over B12N12 and B11N12C nanocages. Two possible reaction pathways can be proposed for the oxidation of CO with O2 molecule over these surfaces: CO + O2 → OOCO → CO2 + Oads and CO + Oads → CO2. Both Eley–Rideal (ER) and Langmuir–Hinshelwood (LH) mechanisms are considered for these two reaction pathways. Our calculations indicate that the CO oxidation reaction over the B11N12C nanocage proceeds via the ER mechanism followed by LH mechanism with an activation energy (Eact) of 0.58 eV. In the case of the B12N12 nanocage, it can be estimated that both reaction pathways go through the LH mechanism. The Eact of the first reaction step is about 2.5 eV, while it is negligible for the second route. Based on the present theoretical results, the catalytic activity of the B11N12C nanocage toward the CO oxidation reaction is more than that of the B12N12 cluster. This can be related to the presence of a C atom that plays a significant role in the activation of the whole cluster. Meanwhile, the performance of the B11N12C nanocage as a catalyst used for the oxidation of CO with O2 molecules may proceeded at near ambient temperatures. These results indicate that the B11N12C nanocage can be utilized as a favorable low-cost catalyst for the CO oxidation reaction.
 Please wait while we load your content...
Please wait while we load your content...

