
Chemo- and regioselective head-to-tail heterodimerization of vinylarenes with 1,1-diphenylethene over a heterogeneous catalyst (Snβ zeolite)†
Abstract
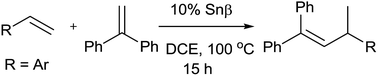
In the presence of Snβ zeolite, vinylarenes undergo the highly chemo- and regioselective head-to-tail heterodimerization with 1,1-diphenylethene to form the corresponding alkenes in good to excellent yields. The scope of the reaction was explored by various vinylarenes with 1,1-diphenylethene. However, the substrates with strong electron-withdrawing groups failed to react with 1,1-diphenylethene under the present catalytic conditions. Moreover, the Snβ zeolite is recyclable and can be reused without significant loss in its catalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

