
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and rheological behavior of poly(1,2-butylene oxide) based supramolecular architectures†
Jürgen
Allgaier
*a,
Claas H.
Hövelmann
a,
Zhang
Wei‡
a,
Mariapaola
Staropoli
a,
Wim
Pyckhout-Hintzen
a,
Nicole
Lühmann
b and
Sabine
Willbold
b
aJülich Centre for Neutron Science JCNS, Institute for Complex Systems ICS, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: j.allgaier@fz-juelich.de
bCentral Institute for Engineering, Electronics and Analytics, ZEA-3: Analytics, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
First published on 8th January 2016
Abstract
In this work we present the synthesis of poly(1,2-butylene oxide) (PBO) functionalized with the complementary hydrogen bond forming groups 2,4-diaminotriazine (DAT) and thymine. PBO is a rubbery polymer. Due to its semi-polar nature PBO is expected to suppresses non-directed cluster formation of the supramolecular groups but not influence their directed interactions. For the synthesis of backbone functionalized polymers we developed a procedure which allowed randomly copolymerizing BO with 1,2-epoxy-7-octene using anionic ring opening polymerization with potassium tert-butanolate as initiator. The vinyl groups were converted to OH-groups by oxidation. In addition, PBO with one alcoholic end group was obtained by homopolymerization of BO. For the variant with OH-groups at both chain ends a procedure was developed which was based on the cleavage of the tert-butyl initiator group. In all the polymers the alcohol groups were basically quantitatively transformed into NH2-groups. DAT and thymine functionalities were attached to the NH2-groups again in almost quantitative conversion. All reaction steps were monitored by 1H-NMR using pyridine-d5 as solvent. This method allowed determining the conversions of the different synthesis steps with high precision. The materials were examined in linear rheology in order to study the effect of the hydrogen-bonds on the dynamics of the resulting supramolecular structures. The results corroborate the exclusive existence of directed interactions between the supramolecular groups.
Introduction
Supramolecular chemistry describes the intermolecular interactions of molecules and has been defined as “chemistry beyond the molecule”.1 Since the 1980s supramolecular interactions have been increasingly utilized in polymer chemistry to create supramolecular polymers2 consisting of macromolecules that interact through directed non-covalent bonds.3 Due to their reversible bonding and ease of self-organization, supramolecular polymers have been extensively used in the construction of complex polymer architectures4 and the design of self-healing polymers.5Main-chain supramolecular polymers consist of smaller molecules that assemble into linear chains through non-covalent interactions. Side-chain supramolecular polymers result from intermolecular attachment of side-chains to a linear polymer backbone resulting in the formation of comb-like supramolecular structures6 or supramolecular networks (Scheme 1).7 While intermolecular interactions can result from multiple forces like π–π interactions, metal–ligand binding or ionic attraction, hydrogen bonding is the most common motif in the construction of supramolecular polymers. The existence of non-directed aggregation renders the description of the underlying models for reversible hydrogen bonding rather complex.
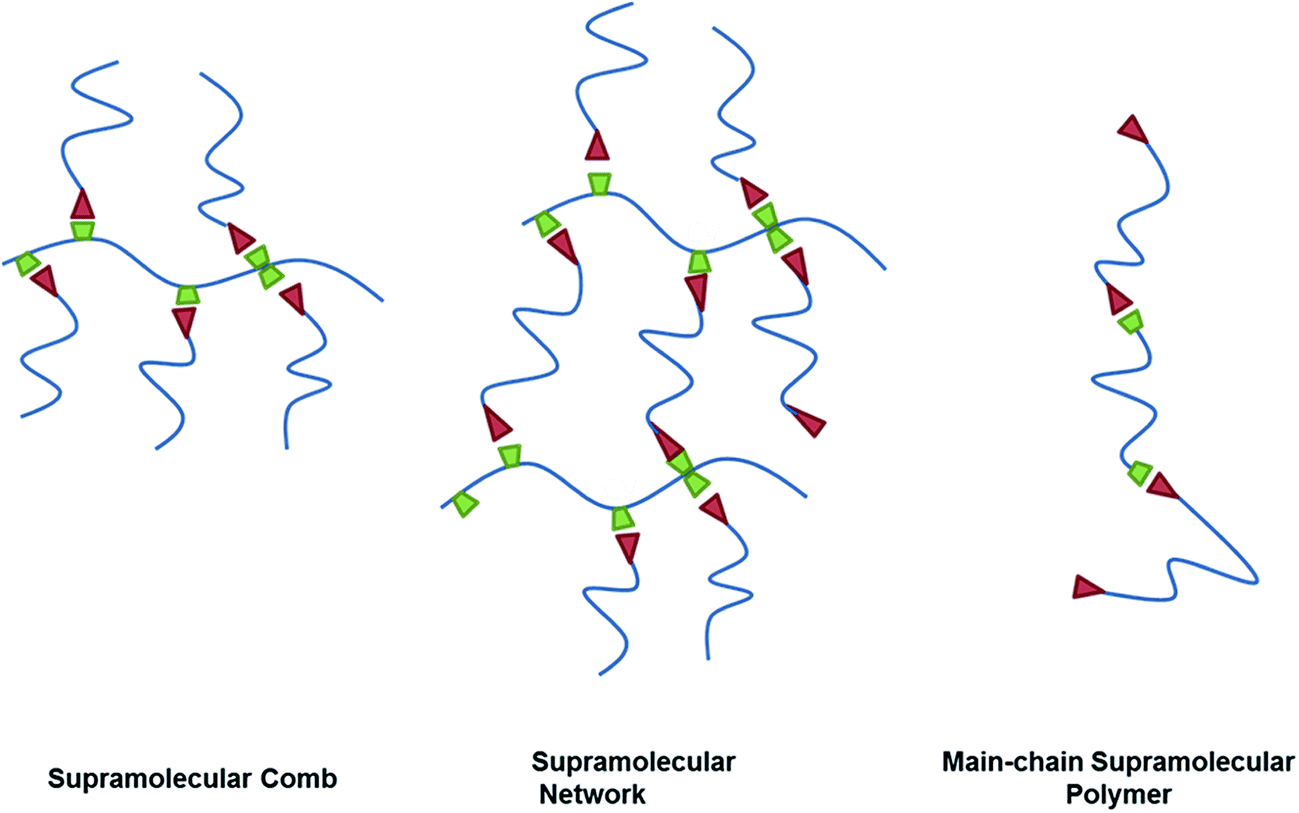 | ||
| Scheme 1 Supramolecular polymer architectures. | ||
To investigate association phenomena and polymer chain dynamics in hydrogen bonding supramolecular polymers we envisioned the construction of a toolkit for the assembly of different supramolecular architectures based on poly(1,2-butylene oxide) (PBO), with hydrogen-bonding groups randomly attached along the backbone or to the chain ends (Scheme 1). 2,6-Diaminotriazine (DAT) and thymine (Thy) are a common motif in the construction of supramolecular structures. They can form strong hetero-associates through three hydrogen bonds8 while the homo-association through two hydrogen bonds shows weaker interaction.
This pair has been previously used in the construction of linear main-chain supramolecular polymers9 but is also ideally suited for the assembly of comb-like polymers10 and polymer networks.11 In solution the two moieties exhibit a high tendency to hetero-associate, which favors the formation of defined supramolecular structures. In small angle neutron scattering measurements (SANS) chain extended structures were observed in solution using polypropylene glycol functionalized with thymine and DAT units.12 Using the comb architecture a complementary association would lead to the formation of a transiently branched structure, which combines e.g. the increased viscosity of branched polymer systems with the flexibility of the supramolecular association. This system can lose its identity by shearing or temperature increase. This allows higher processability compared to a covalently branched polymer. The disconnected structures can reassemble and restore the properties to reobtain the branched structure.
In the melt state coordination can differ substantially from the behavior in solution. The aggregation of thymine and DAT groups in a polymer melt seems to depend highly on the nature of the involved polymer and possibly even the method of attaching the groups to the polymer chains. The thymine/DAT pair has been used with a variety of polymers in a large range of polarities. End-group functionalized PE9d shows phase separation between the polar end-groups and the non-polar polymer. In PIB functionalized with thymine/DAT along the backbone11 or at the chain-ends,9a spherical micelles, consisting of thymine–DAT aggregates were detected in small angle X-ray scattering (SAXS) measurements in the melt state.13 Moving to more polar polymers a detailed study by Herbst and Binder shows strong self-association of the DAT groups in the melt in telechelic PnBA.14 The complementary thymine–DAT association is shown to be of a similar strength but does not seem to result from hydrogen bonding thereby differing considerably from the scenario in solution. Similarities to the association in solution have only been observed when a polar polymer like PPO9b or PMMA15 is employed. While lamellar phases are observed in thymine functionalized telechelic PPO at ambient temperature, in the combination with DAT the lamellar structures disappeared.9c At higher temperature an increasing association DAT–DAT < Thy–Thy ≪ DAT–Thy is observed. In order to obtain a molecular understanding of the interactions in the bulk state, systems are required where only directed interactions exist between the supramolecular groups and not non-directed interactions leading for example to cluster formation. The latter scenario i.e. a microphase separation of supramolecular groups in the melt preferably occurs if non-polar polymers are used. Strongly polar polymers, on the other hand, substantially weaken the supramolecular interactions.
PBO is a moderately polar polymer that is miscible with polar solvents like ethanol. The glass transition temperature is about −70 °C and the polymer is equipped with a reasonably low entanglement molecular weight of 8.800 g mol−1.16 In contrast to PPO, PBO can be polymerized to high molecular weights with narrow molecular weight distributions using anionic ring opening polymerization.18 In this work we present new synthetic techniques to obtain PBO being randomly functionalized with DAT or thymine units along the backbone or alternatively at the chain ends. We developed a method to functionalize PBO with alcohol and subsequently with amino groups. Commercially available DAT and thymine precursors were connected to the PBOs using classical esterification or amidation reactions. This avoids the use of CuAAC click chemistry9a and the associated difficulties to remove the copper catalyst. In addition, we outline the detailed examination of the polymers using 1H-NMR in pyridine-d5. This technique turned out to be ideal to precisely determine functionalization degrees. This knowledge is especially important for chain end functionalized polymers, which form a supramolecular condensation polymer. Similar to polycondensation reactions non-functional chain ends drastically lower already in small concentrations the supramolecular polymerization degree. The materials were examined in linear rheology, in order to study the effect of the hydrogen-bonds on the dynamics of the resulting supramolecular structures.
Experimental section
General techniques
All synthetic manipulations were carried out in a glove box, filled with argon (M Braun, Unilab) or at a high vacuum line using standard cryogenic distillation techniques. Air and moisture sensitive agents were handled in flasks equipped with Teflon stopcocks that allowed transferring materials between vacuum line and glove box without contamination with air. The flasks were pressure tested to 4 to 12 bars, depending on the size of the flask. Polymers were purified by washing the solvent free raw products several times with methanol or methanol–water mixtures under stirring. Water was added for the low molecular weight polymers to reduce their solubility in methanol.Materials
Toluene (Merck, ≥99.9%) was degassed, distilled into another flask which contained sodium metal, stirred over the sodium for at least 24 hours before being degassed again and heated to 110–115 °C for 3–4 hours. THF (KMF, ≥99.9%) was degassed, pre-dried over CaH2 and then distilled into a flask containing potassium metal and benzophenone. Dichloromethane (Sigma Aldrich, ≥99.8%) was dried over CaH2. Triethylamine (Sigma Aldrich, ≥99%) was degassed and distilled into a flask containing solvent-free n-butyllithium. After the drying procedures the solvents were distilled into storage flasks equipped with Teflon stopcocks. Methanesulfonyl chloride (Fluka, ≥98%) was distilled into another flask without additional drying step.1,2-Butylene oxide (BO) (Sigma Aldrich, lot purity 99.9%) was first degassed and then stirred over CaH2 for 4 days. This process was repeated first again at room temperature and then at 60 °C. 1,2-Epoxy-5-hexene (EH) (Sigma Aldrich, lot purity 99.4%) was treated in the same way but executing the last two drying step at 80 °C and 100 °C. 1,2-Epoxy-7-octene (EOc) (Alfa Aesar, lot purity 98%) and 1,2-epoxy-9-decene (ED) (Alfa Aesar, lot purity 96%) were first fractionally distilled at a pressure of 60 mbar using a distillation apparatus, equipped with a 30 cm column filled with wire mesh rings. For EOc the fraction distilling between 85.5 and 86 °C and for ED the fraction distilling between 121 and 121.5 °C was used for the drying process over CaH2. This procedure was carried out as described for BO except that the second and the third drying steps were carried out at 100 °C. All monomers were distilled into storage flasks equipped with Teflon stopcocks and kept in the glove box.
Potassium tert-butanolate (KOt-Bu) (Sigma-Aldrich, 99%) was used as received. The crown ether 18-crown-6 (18C6) (Sigma-Aldrich, 99%) was freeze dried with benzene prior to use under high vacuum conditions. The following compounds were used as received: 9-borabicyclo[3.3.1]nonane (9-BBN) solution (Sigma Aldrich, 0.46 M in THF), hydrogen peroxide solution (Sigma Aldrich, 30%), triisopropylsilane (Sigma Aldrich, 99%), trifluoroacetic acid (Sigma Aldrich, 99%), sodium methoxide (Sigma Aldrich, 95%), azidotrimethylsilane (Sigma Aldrich, 95%), tetrabutylammonium fluoride (TBAF) solution (Sigma Aldrich, 1.0 M in THF), lithium aluminium hydride solution (Sigma Aldrich, 2.1 M in THF), N,N-dimethylformamide (DMF) (Sigma Aldrich, over molecular sieve, ≥99.8%), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) (Sigma Aldrich, ≥97%), N,N-diisopropylethylamine (Sigma Aldrich, ≥99%), thymine-1-acetic acid (Sigma Aldrich, 98%), N,N-dimethylacetamide (DMA) (Sigma Aldrich, ≥99.9%), 2-chloro-4,6-diamino-1,3,5-triazine (DAT-Cl) (Sigma Aldrich, 98%).
Synthesis of PBO5k1-OH and PBO5k2-OH18
The BO homopolymerization reactions were carried out in flasks equipped with Teflon stopcocks. The flasks were filled inside the glove box with KOt-Bu and 18C6. Precise weight control was achieved by the use of an analytical balance which was installed in the glove box. Then dry toluene was added. The pre-determined amount of BO was distilled into the reaction flasks at the vacuum line. The molar ratio of 18C6 and KOt-Bu was 0.50 and the mass ratio of BO to toluene was about 1. In a typical polymerization run (sample PBO5k1-OH) KOt-Bu (0.339 g, 3.02 mmol), 18C6 (0.398 g, 1.51 mmol), 15.14 g of BO and 16 mL of toluene were used. The reaction mixture was left for about 6 days at −15 °C and was terminated with acetic acid. After this all volatile materials were removed by vacuum distillation and the polymer was dissolved in pentane in order to remove most of the potassium acetate by centrifugation. After the removal of pentane the polymer was stirred twice with mixtures of methanol and water (mass ratio 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and was dried under high vacuum with stirring.
:1) and was dried under high vacuum with stirring.
Synthesis of P(BO/EH)11k-OH, P(BO/EOc)11k-OH, P(BO/ED)12k-OH and P(BO/EOc)38k-OH
The experiments for determining the copolymerization behavior of samples P(BO/EH)11k-OH, P(BO/EOc)11k-OH and P(BO/ED)12k-OH were carried out at −10 °C at a molar ratio of 18C6 to KOt-Bu of 0.9 to 1.0 and a mass ratio of monomer to toluene of about 1. The pre-cooled monomer mixtures were added to the pre-cooled mixtures of KOt-Bu, 18C6 and toluene inside the glovebox. The reaction flasks equipped with Teflon stopcocks were stored in a refrigerator inside the glove box. Samples were taken at different times and immediately terminated with acetic acid. After drying under high vacuum conditions the conversions were calculated from the weights of the non-volatile residues. In a typical polymerization run (sample P(BO/ED)12k-OH) KOt-Bu (0.225 g, 2.01 mmol), 18C6 (0.526 g, 1.99 mmol), BO (15.86 g, 220 mmol), ED (8.51 g, 55 mmol) and 21.7 g of toluene were used and samples were taken after 70 min, 3.2 h, 5.2 h, 8.2 h and 25.8 h. For sample P(BO/EOc)38k-OH KOt-Bu (0.072 g, 0.64 mmol) and 18C6 (0.336 g 1.27 mmol) were used so that the molar ratio of 18C6 to KOt-Bu was 2.0. BO (28.55 g, 396 mmol), EOc (0.857 g, 6.8 mmol) and 29.58 g of toluene were added and samples were taken after 70 min, 3.2 h, 20.5 h and 68 h. The final product was washed with methanol and was dried under high vacuum conditions.Synthesis of PBO38k(OH)8(OH)17
P(BO/EOc)38k-OH (12.263 g, 2.83 mmol vinyl groups) was dissolved in 63.3 g of dry THF in a flask equipped with a Teflon stopcock. The concentration of vinyl groups was determined separately by 1H-NMR using 1,1,1,2,2-pentabromoethane as internal standard to be 0.231 mmol g−1. The polymer solution was cooled to −15 °C and 9-BBN solution (6.8 mL, 3.13 mmol) was added inside a glovebox. The reaction mixture was left for 0.5 h at −15 °C, 2 h at −10 °C and overnight at 0 °C. After warming up to room temperature 1 mL of degassed methanol was added. After cooling to −25 °C 0.727 g of an aqueous sodium hydroxide solution containing 3.623 mmol NaOH and 1.075 g of H2O2 solution containing 9.379 mmol of H2O2 were added under argon atmosphere. The exact concentration of the H2O2 solution was determined before by iodometric titration. The reaction mixture was allowed to warm up to room temperature overnight. After partial removal of the solvent under reduced pressure the product mixture was first washed three times with water and twice with a mixture of methanol and water (mass ratio 1:1). Finally the product was dried under high vacuum conditions.
Synthesis of HO-PBO5k-OH
15.56 g of PBO5k2-OH was dissolved in 400 g of CH2Cl2. Then triisopropylsilane (3.904 g, 24.7 mmol) and trifluoroacetic acid (62.26 g, 546 mmol) were added. After stirring at 40 °C for about 7 h the mixture was cooled to 0 °C. All volatile materials were distilled off and the obtained product TFA-PBO5k-TFA was stirred under high vacuum conditions overnight. A small sample was taken for characterization and all residual material was dissolved in 200 mL of dry THF inside a glove box, followed by the addition of sodium methoxide (5.35 g, 99 mmol). The suspension was stirred for 3 h. 1 L CH2Cl2 was added and the organic phase was washed 4 times with water. The solvent was removed under reduced pressure and the obtained raw product was washed twice with a mixture of methanol and water (mass ratio 3:1). After drying under high vacuum conditions 14.42 g of product was obtained.
Synthesis of PBO5k-N3, N3-PBO5k-N3 and PBO38k(N3)8(N3)
For the synthesis of PBO5k-N3 PBO5k1-OH (3.08 g, 0.64 mmol OH) was dissolved in 70 g of dry CH2Cl2 inside a glove box. Dry triethylamine (2.552 g, 25.2 mmol) was added and the mixture was cooled to −10 °C. Then methanesulfonyl chloride (1.623 g, 14.2 mmol) was added under strong stirring. The mixture was allowed warming up to room temperature. After one day the mixture was dark orange and after three days volatile materials were removed at a vacuum line, followed by additional pumping for 3 h. THF (45 mL) was added and the remaining solid material was removed by centrifugation. Then azidotrimethylsilane (1.584 g, 13.7 mmol) and TBAF solution (13.5 mL, 13.5 mmol) were added. The mixture was stirred at 50 °C for 67 h and was then filtered through silica gel. The silica gel was rinsed with 250 mL of THF. After the removal of THF under reduced pressure 250 mL of chloroform was added and the solution was washed 3 times with water. The solvent was again removed under reduced pressure and the viscous raw product was washed 3 times with a mixture of methanol and water (mass ratio 8:1) and finally dried under high vacuum conditions. 2.71 g of product was obtained. For the synthesis of N3-PBO5k-N3 HO-PBO5k-OH (12.96 g, 5.08 mmol OH) was used together with 350 g of CH2Cl2, triethylamine (22.011 g, 218 mmol) and methanesulfonyl chloride (13.924 g, 122 mmol). The reaction was left for 3 days at room temperature. The mesylate intermediate product was dissolved in 220 mL of THF, was centrifuged and azidotrimethylsilane (13.565 g, 118 mmol) together with TBAF solution (116 mL, 116 mmol) were used for the azide formation. After 4 days at 50 °C the product was purified as described for PBO5k-N3 and 9.82 g of product was obtained. PBO38k(N3)8(N3) was synthesized using PBO38k(OH)8(OH) (1.95 g, 0.47 mmol OH) together with 70 g CH2Cl2, triethylamine (6.44 g, 63.6 mmol) and methanesulfonyl chloride (3.62 g, 31.6 mmol). The reaction was left overnight at room temperature. The mesylate intermediate product was dissolved in 55 mL of THF, was centrifuged and azidotrimethylsilane (0.873 g, 7.58 mmol) together with TBAF solution (7.5 mL, 7.5 mmol) were used for the azide formation. After one day at 50 °C the product was purified as described for PBO5k-N3. The final washing procedure was carried out using only methanol and 1.57 g of product was obtained.
Synthesis of PBO5k-NH2. NH2-PBO5k-NH2 and PBO38k(NH2)8(NH2)
PBO5k-NH2 was synthesized using PBO5k-N3 (2.638 g, 0.55 mmol N3) which was dissolved in 20 mL of dry THF inside a glove box. LiAlH4 solution (2.9 mL, 6.09 mmol) was added at room temperature under stirring. After two days 1.4 mL of a saturated aqueous NH4Cl solution was added slowly to the pre-cooled reaction mixture. Solid material was removed by centrifugation and after removal of the solvent the raw product was washed 3 times with a mixture of methanol and water (mass ratio 9:1) and finally dried under high vacuum conditions and 2.31 g of product was obtained. NH2-PBO5k-NH2 was synthesized using N3-PBO5k-N3 (9.65 g, 3.74 mmol N3) which was dissolved in 95 mL dry THF and reacted with LiAlH4 solution (22.5 mL, 47.3 mmol). The excess hydride was quenched with 11.3 mL of saturated NH4Cl solution and the raw product was washed with a mixture of methanol and water (mass ratio 4:1). After drying under high vacuum conditions 9.32 g of product was obtained. PBO38k(NH2)8(NH2) was synthesized with PBO38k(N3)8(N3) (1.524 g, 0.37 mmol N3) dissolved in 20 mL of dry THF. LiAlH4 solution (2.0 mL, 4.2 mmol) was added and after 5 min a clear gel formed. Prior to the quenching with 1.0 mL of saturated NH4Cl solution extra THF was added. The raw product was finally washed in pure methanol. After drying under high vacuum conditions 1.52 g of product was obtained.
Synthesis of PBO38k(Thy)8(OH)
PBO38k(OH)8(OH) (6.79 g, 1.64 mmol OH) was dissolved inside a glove box in a mixture of 16.3 g of dry THF and 32.4 g of dry DMF. TBTU (0.850 g, 2.65 mmol), N,N-diisopropylethylamine (1.09 g, 8.43 mmol) and thymine-1-acetic acid (0.442 g, 2.40 mmol) were added. After 10 min of stirring at room temperature solid material precipitated. The reaction was continued overnight and the precipitate was removed by centrifugation. After removal of the solvent the raw product was first washed 3 times with 0.2 M aqueous NaHCO3, followed by a mixture of methanol and water (mass ratio 1:1) and finally pure methanol. After drying under high vacuum conditions 6.12 g of product was obtained.
Synthesis of PBO5k-Thy and Thy-PBO5k-Thy
PBO5k-Thy was synthesized using PBO5k-NH2 (0.52 g, 0.11 mmol NH2) which was dissolved in a mixture of 1.05 g of dry THF and 1.01 g of dry DMF inside a glove box. Then TBTU (0.085 g, 0.27 mmol), N,N-diisopropylethylamine (0.079 g, 0.61 mmol) and thymine-1-acetic acid (0.037 g, 0.20 mmol) were added. The mixture was stirred for 41 h at 40 °C. Then the solvent was removed at a vacuum line and the raw product was dissolved in 10 mL of pentane. The solid material which already was formed during the reaction was removed by centrifugation. After removal of the pentane the polymer was first washed 3 times with 0.2 M aqueous NaHCO3, followed by a mixture of methanol and water (mass ratio 1:1). After drying under high vacuum conditions 0.54 g of product was obtained. Thy-PBO5k-Thy was synthesized using NH2-PBO5k-NH2 (2.311 g, 0.91 mmol NH2), dissolved in 4.87 g of dry THF and 4.88 g of dry DMF. Then TBTU (0.759 g, 2.37 mmol), N,N-diisopropylethylamine (0.720 g, 5.57 mmol) and thymine-1-acetic acid (0.334 g, 1.81 mmol) were added and the reaction was performed as described for PBO5k-NH-Thy. After drying 2.177 g of product was obtained.
Synthesis of PBO5k-DAT, DAT-PBO5k-DAT and PBO38k(DAT)8(DAT)
PBO5k-DAT was synthesized using of PBO5k-NH2 (1.650 g, 0.34 mmol NH2). In a flask equipped with a Teflon stopcock 7.0 g of DMA were added together with NaHCO3 (0.074 g, 0.88 mmol) and DAT-Cl (0.088 g, 0.60 mmol). The mixture was carefully degassed several times at a vacuum line and heated to 100 °C for 7 days. After removal of the solvent under high vacuum conditions 20 mL of pentane was added and the solid material was removed by centrifugation. The pentane was removed and the raw product was washed 3 times with a mixture of DMF and water (mass ratio 9:1). After the drying procedure under high vacuum conditions 1.191 g of product was obtained. DAT-PBO5k-DAT was synthesized using NH2-PBO5k-NH2 (5.483 g, 2.15 mmol NH2), NaHCO3 (0.525 g, 6.45 mmol) and DAT-Cl (0.626 g, 4.30 mmol) and 28.2 g of DMA. The mixture was stirred for 10 days at 100 °C. The purification process was similar to PBO5k-DAT and after the drying procedure 4.288 g of product was obtained. PBO38k(DAT)8(DAT) was synthesized using PBO38k(NH2)8(NH2) (1.36 g, 0.32 mmol NH2), NaHCO3 (0.089 g, 1.06 mmol) and DAT-Cl (0.131 g, 0.90 mmol) and 8.1 g of DMA. The mixture was left for 4 days at 80 °C. The purification process was similar to PBO5k-DAT and after the drying procedure 1.09 g of product was obtained.
Polymer characterization
Size exclusion chromatography (SEC) experiments were carried out using a Polymer Laboratories PL 220 SEC instrument equipped with a differential refractive index detector and with three PolyPore columns at 50 °C. The solvent was a mixture of THF and DMA (85:15 by volume) at a flow rate of 1 mL min−1 polystyrene standards were used for calibration. Some polymers were additionally examined using an Agilent SEC instrument equipped with a differential refractive index detector, a Wyatt DAWN HELEOS 8+ light scattering detector and three PolyPore columns at 30 °C. The solvent was THF at a flow rate of 1 mL min−1. NMR spectra were collected on a Bruker Avance III 600 MHz spectrometer equipped with a Prodigy cryoprobe or on a Varian Inova 400 MHz with a 5 mm PFG AutoX DB probe. Samples of the polymers were dissolved in either in CDCl3 or in pyridine-d5 and measured at 295 K. Infrared spectra were collected on a Bruker Tensor 27 equipped with a Golden Gate diamond attenuated total reflection unit.
Linear rheology
PBO38k(OH)8(OH), PBO38k(Thy)8(OH), PBO5k-DAT and PBO5k1-OH were investigated in small amplitude oscillatory shear in a parallel plate mode on an ARES rheometer (Rheometrics Sci.) equipped with a 2K-FRNT transducer. The diameter of the plates was 25 mm and the sample gap about 1 mm. Isothermal frequency sweeps were performed in the range between 0.1 < ω < 100 rad s−1 and a temperature range between −50 < T < 0 °C under a nitrogen blanket to avoid water condensation. The data were converted into master curves at a reference temperature T0 = −25 °C for PBO38k(OH)8(OH) and PBO38k(Thy)8(OH) and −35 °C for PBO5k-DAT and PBO5k1-OH using the TTS procedure. The shift factor aT was determined as logaT = −C1(T − T0)/(C2 + (T − T0)). The obtained WLF parameters for PBO38k(OH)8(OH) were C1 = 6.79 and C2 = 93.67 °C and for PBO38k(Thy)8(OH) C1 = 7.23 and C2 = 97.75 °C. The WLF parameters at −35 °C for PBO5k1-OH were C1 = 6.05 and C2 = 66.10 °C and for PBO5k-DAT C1 = 10.29 and C2 = 88.46 °C. Tg values of the polymers were determined by DSC (Q2000, TA Instruments) at heating and cooling rates of 10 °C min−1 to be −66 ± 1 °C (PBO38k(OH)8(OH)) and −61 ± 1 °C (PBO38k(Thy)8(OH)), −65 ± 1 °C PBO5k-DAT and −68 ± 1 °C PBO5k1-OH.
Results and discussion
For the functionalization of PBO with supramolecular groups our objective was to use preferably commercially available precursors. We have chosen the DAT/thymine pair as complementary hydrogen bond forming system. A DAT precursor is commercially available in high purity as 2-chloro-4,6-diamino-1,3,5-triazine (DAT-Cl) and can be attached to polymer chains via nucleophilic aromatic substitution by the amine end-groups of the polymer.9b The same scenario holds for thymine-1-acetic acid which in addition can be attached directly to alcohol groups of the polymer. Therefore the synthetic strategy comprised first the production of alcohol functionalized polymers and subsequent transformation of these groups into amino groups.End-functionalized polymers equipped with alcohol and amino groups
PBO with narrow molecular weight distribution can be obtained by anionic ring opening polymerization of BO monomer at low temperatures in the presence of crown ethers.18 In our polymerization experiments we use potassium tert-butanolate as initiator. After acidic termination this yields polymers with initial tert-butoxy units and terminal secondary alcoholic groups (see Scheme 2). Fig. 1 shows the NMR spectrum of sample PBO5k1-OH in CDCl3. The molecular weight characterization is given in Table 1.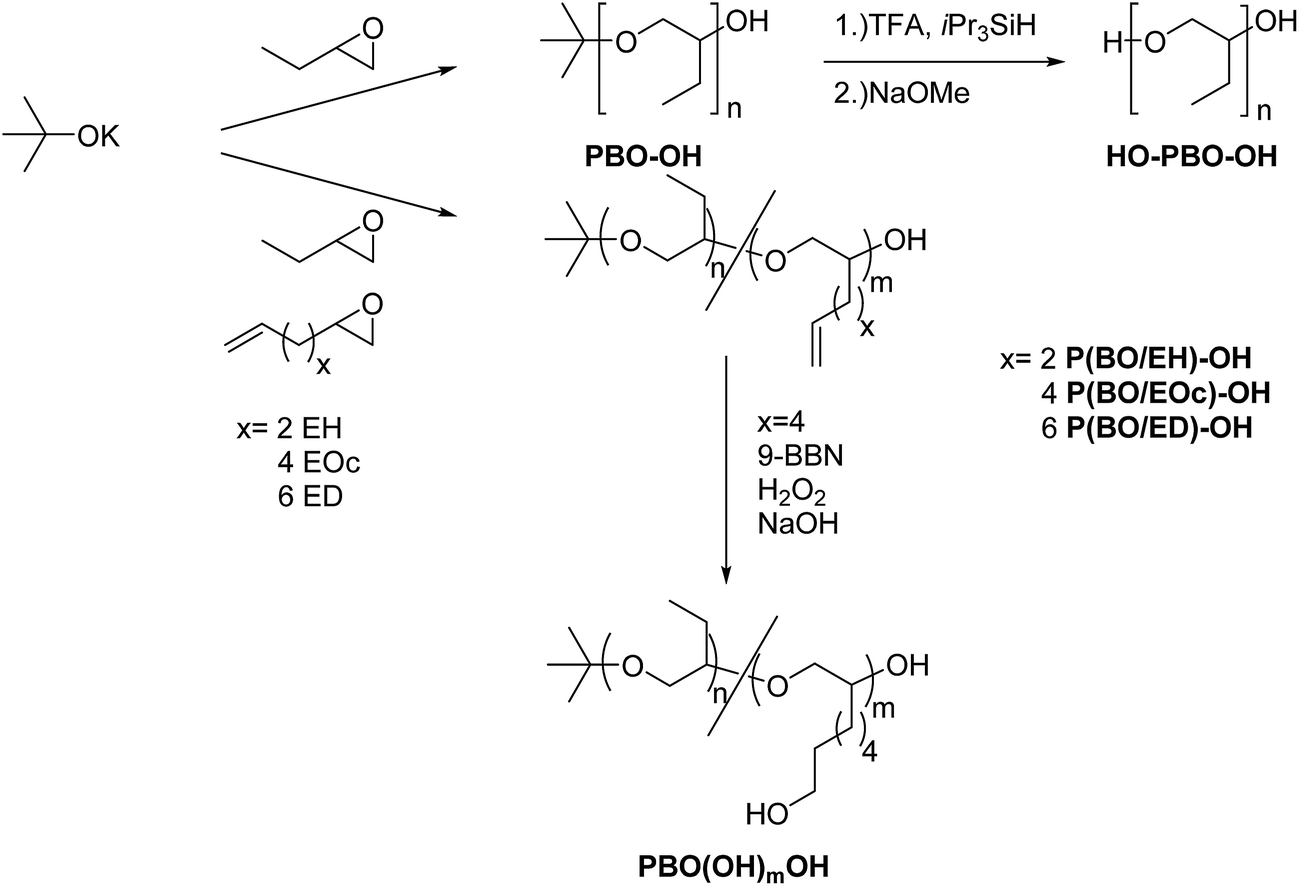 | ||
| Scheme 2 Synthesis of OH-functionalized poly(1,2-butylene oxide)s. | ||
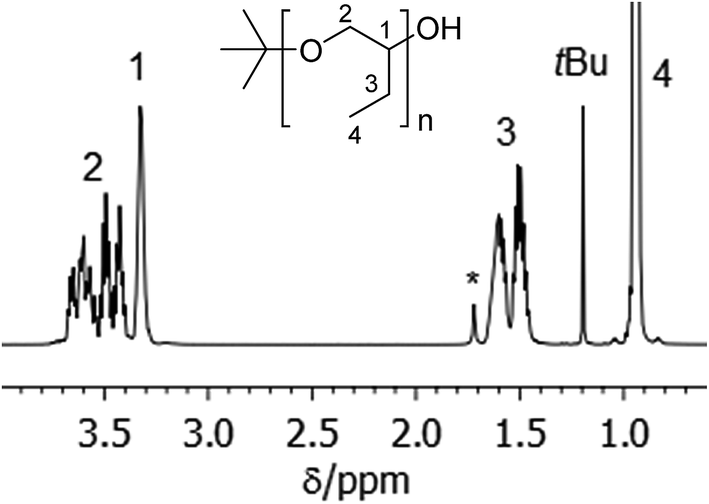 | ||
| Fig. 1 1H-NMR spectrum in CDCl3 of PBO5k1-OH. The signal marked with * belongs to water, which is shifted to lower fields in the presence of polyethers. | ||
| M n (NMRa) | M n (SEC/LS) | M w/Mn (SEC) | Mass fraction of coupled chains (SEC) | |
|---|---|---|---|---|
| a Calculated from the signal intensities of the tert-butyl initiator signal and the polymer signals from the 1H-NMR spectra in CDCl3. | ||||
| Monofunctional polymers | ||||
| PBO5k1-OH | 4.810 | 4.400 | 1.03 | 0% |
| PBO5k-N3 | 4.820 | 1.04 | <1% | |
| PBO5k-Thy | 4.950 | 1.03 | 1% | |
| PBO5k-DAT | 4.870 | 1.06 | 5% | |
|
||||
| Bifunctional polymers | ||||
| PBO5k2-OH | 5.160 | 1.02 | 0% | |
| HO-PBO5k-OH | 5.070 | 1.03 | 2% | |
| N3-PBO5k-N3 | 5.150 | 1.03 | 4% | |
| Thy-PBO5k-Thy | 1.04 | 4% | ||
| DAT-PBO5k-DAT | 1.08 | 12% | ||
|
||||
| Side chain functionalized polymers | ||||
| P(BO/EOc)38k-OH | 38.400 | 34.200 | 1.02 | 0% |
| PBO38k(OH)8(OH) | 37.400 | 35.000 | 1.02 | 1% |
| PBO38k(N3)8(N3) | 36.900 | 35.900 | 1.02 | 1% |
| PBO38k(Thy)8(OH) | 40.600 | 1.03 | 2% | |
| PBO38k(DAT)8(DAT) | 1.62 | 25% | ||
Bifunctional polymers containing OH-groups at both chain ends can be obtained using diol based initiators. At the low polymerization temperatures, needed to suppress side reactions in the polymerization of BO, fully metalated initiators are required. However, alkali metal dialcoholates are insoluble in common organic solvents. Therefore we used an initiator with a protected hydroxyl function for the synthesis of the bifunctional polymers. Surprisingly, it turned out that potassium tert-butanolate is not only a versatile initiator for monofunctional polymers but the initial tert-butoxy unit additionally can be used as protective group. The cleavage of the initial tert-butoxy unit of PBO5k2-OH was carried out using trifluoroacetic acid and triisopropylsilane as scavenger for the intermediately formed tert-butyl cations (Scheme 2).19 The trifluoroacetate diester TFA-PBO5k-TFA was obtained as product of this first step after a reaction time of 7 h at 40 °C. After subsequent cleavage of the ester by treatment with sodium methoxide20 to the diol product HO-PBO5k-OH no remaining TFA-ester could be detected by NMR.
The molecular weight characterization data of HO-PBO5k-OH are listed in Table 1 and the SEC trace together with the one of the mother compound PBO5k2-OH is given in Fig. 2. They are identical in the low molecular weight region. Therefore chain scission reactions via the cleavage of inner ether bonds can be ruled out. The small signal in the SEC trace of HO-PBO5k-OH at about 22.4 mL represents approximately 2% of the material and corresponds to the double molecular weight compared to the main polymer signal. The high molecular weight material was not present in TFA-PBO5k-TFA nor could it be detected in other similar experiments. There is literature precedence for benzylic trifluoroacetates as nucleofuge in SN1-type reactions21 which could explain the dimerization. However, it seems unlikely that this reaction would occur at trifluoroacetate esters of the aliphatic alcohol chain ends. Despite the high MW impurity the molecular weight distribution stayed narrow (Table 1). While the use of the tert-butoxy unit as protective group is known for ethylene oxide oligomers20 it has not been used for secondary polyethers like PPO or PBO. The selectivity of the ether cleavage in ethylene oxide based compounds can be expected due to the much lower reactivity of primary ethers compared to the tertiary ether of the tert-butyl end-group. However this selectivity is far less pronounced in the more reactive secondary polyethers present in PBO.22 Unexpectedly, the ether bonds between the monomer units were not attacked and selective cleavage of the tert-butyl end-group could be achieved. The scenario did not change even if the reaction time was increased from one day to several days and higher molecular weight polymers were used.
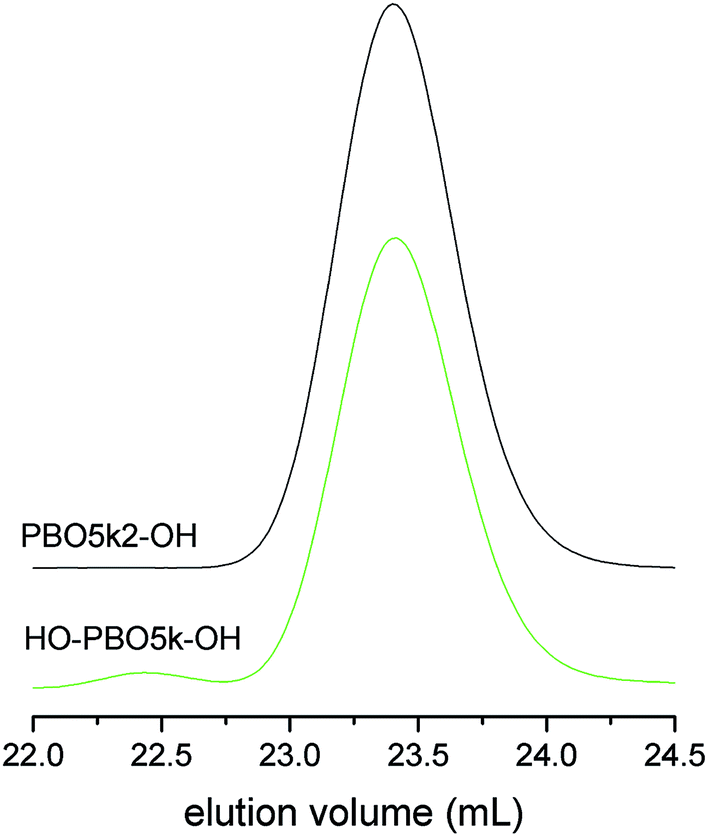 | ||
| Fig. 2 SEC chromatograms of PBO5k2-OH and HO-PBO5k-OH. | ||
The conversion of the alcoholic end-groups to amino groups was carried out in the next step (Scheme 3). First the OH-groups were converted into the mesylate ester using methanesulfonyl chloride followed by reaction with azidotrimethylsilane. Tetrabutylammonium fluoride was added to the reaction to increase the nucleophilicity of the azide in a postulated pentacoordinated silicate intermediate. These reaction conditions were shown to result in increased yields of azidination in secondary halides and tosylates.23 Common azidination procedures using sodium azide in dry DMF24 were not applicable for use with PBO due to the low solubility of the polymer. We therefore used azidotrimethylsilane as azide source due to its increased solubility under less polar conditions. The azide intermediates were subsequently reduced to the amine products using lithium aluminium hydride.
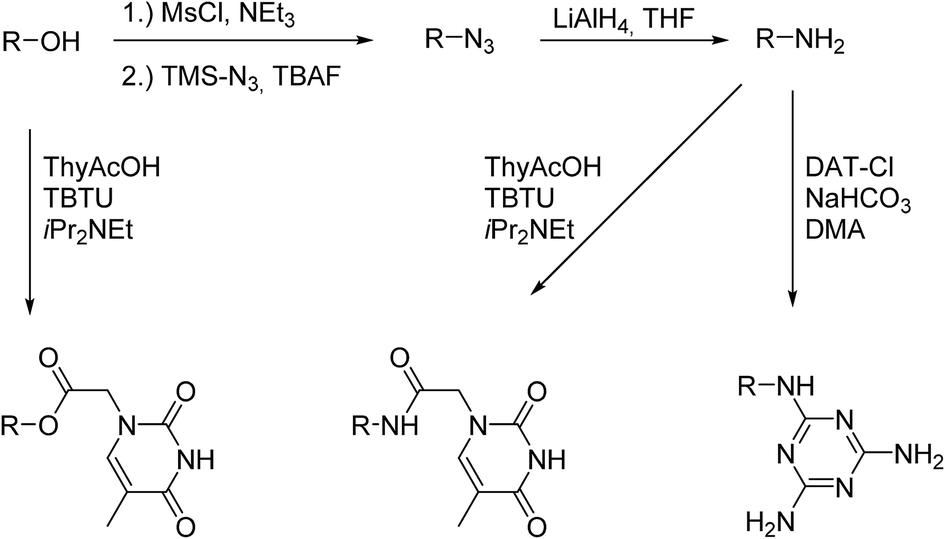 | ||
| Scheme 3 Synthesis of NH2, thymine and DAT functionalized poly(1,2-butylene oxide)s. | ||
NMR spectroscopy is a widely used method for the determination of conversions in organic reactions but cannot be easily adapted for the quantification of polymer functionalizations. The detailed analysis of the conversion in polymer reactions is often difficult due to the high ratio of polymer signals to functional group signals in spectroscopic methods. In addition the NMR signals of the polymer end-groups which are involved in the functionalization are often hidden within the signals of the inner polymer units. Thus unreacted polymer can often not be detected by NMR which renders the quantification of a functionalization reaction difficult. Alternative methods like e.g. MALDI-TOF-MS have been used to determine the degree of functionalization in end-functionalized polymers. However, the ionizability of the starting material can differ from the one in the products which makes it difficult to determine exact conversions. We solved this problem by the use of pyridine-d5 as NMR solvent. In pyridine solutions most of the polyalkylene oxides we investigated showed a high chemical shift of protons at the polymer ends compared to the inner polymer units likely resulting from increased hydrogen bonding of the OH or NH2 end groups with pyridine.
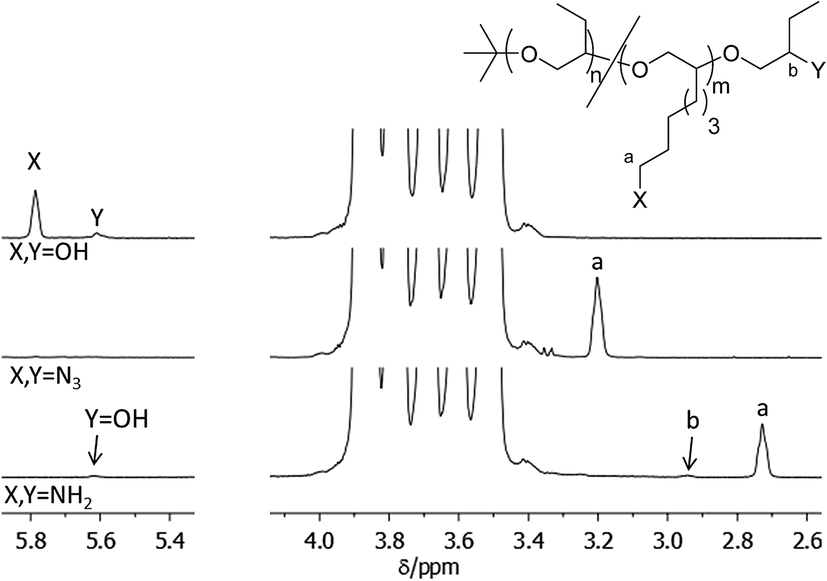
The power of this method can be exemplified in an NMR analysis of the alcohol to azide to amine conversion sequence of HO-PBO5k2-OH (Fig. 3). The OH end-group signals at 5.6 ppm and 5.9 ppm show defined signals in pyridine-d5 with no H/D exchange observed over time. They disappear completely upon conversion to the azide N3-PBO5k-N3. With some substrates however small quantities of alcohol were visible after reduction of the azide to the amine. This can be explained if we assume a quantitative conversion of the alcohol groups to the mesylate intermediate but an incomplete substitution of the mesylate by the azide. The alcohol was then first released in the alkaline workup of the amine product.
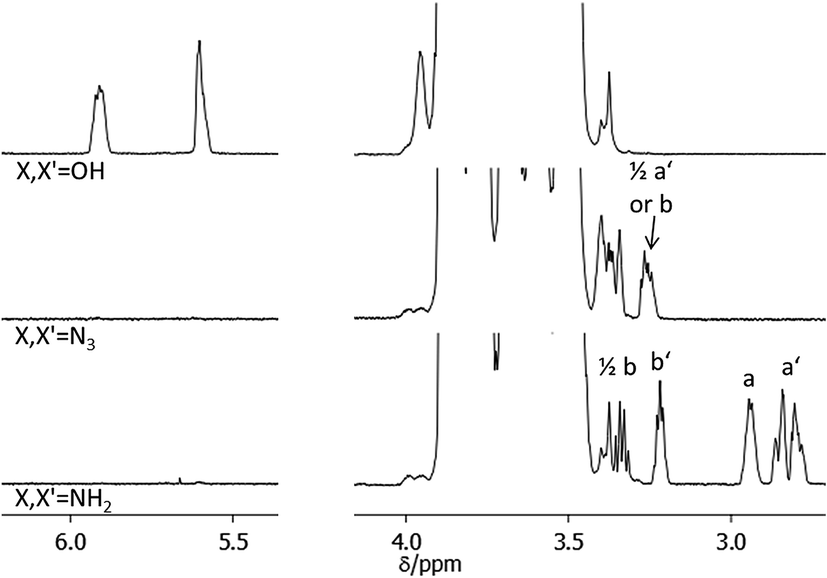 | ||
| Fig. 3 1H-NMR spectrum in pyridine-d5 of the end-group functionalization sequence of HO-PBO5k-OH. | ||
The diastereotopic methylene unit a′ and b in both the primary and secondary PBO ends result in four NMR signals, one of which is visible at 3.3 ppm. Further conversion into the amine form H2N-PBO5k-NH2 results in a NMR spectrum which shows separated signals for three of the diastereotopic protons a′ and b, as determined by H,H-COSY, and both methine protons a and b′. In addition IR measurements were carried out to control the azide reduction. The signal of the azide band at 2095 cm−1 is clearly visible in the spectrum of N3-PBO5k-N3 and fully disappeared in the spectrum of H2N-PBO5k-NH2 (see ESI†). In both conversions no starting material was detected in the product spectra. A similar detailed analysis for the conversions in monofunctional PBO and the functionalization with thymine and DAT can be found in the ESI.†
Side chain functionalized polymers equipped with alcohol and amino groups
Backbone OH-functionalized polymers were synthesized by copolymerizing BO with 1,2-epoxy-ω-alkenes followed by oxidation of the vinyl groups. To achieve a random monomer distribution in a polymerization reaction similar reaction rates of the monomers are required which results in a steady monomer to co-monomer ratio during the polymerization reaction. Kinetic measurements were therefore conducted during the polymerization of BO with the co-monomers EH, EOc and ED (see Scheme 2). In initial experiments the overall monomer to initiator ratio was fixed to about 140 and the mol-fraction of co-monomer was 20%. During the polymerization reactions samples were taken and monomer conversions were determined gravimetrically. The monomer compositions were calculated from the 1H-NMR spectra in CDCl3 by comparing the vinyl signals of the co-monomers at 4.9 ppm and 5.8 ppm with the methyl signal of BO at 0.93 ppm. The results for samples P(BO/EH)11k-OH, P(BO/EOc)11k-OH and P(BO/ED)12k-OH are summarized in Fig. 4 where the cumulative co-monomer fraction is plotted against the overall monomer conversion. The dashed line represents the gravimetrically determined co-monomer fraction of the initial reaction mixture which was identical for all three experiments. Interestingly, EH polymerizes visibly faster than BO whereas for ED the scenario is reverse. However in the case of EOc the co-monomer fraction stays constant over the whole polymerization time indicating a fully random distribution of vinyl groups along the polymer backbone. The only alternative monomer distribution explaining this polymerization behavior would be a blocky structure of the polymer chains. This scenario would require reactivity ratios of the homopolymerization to cross-over reaction step being both ≫1. This is unlikely because of the chemical similarity of both epoxides. The polymerization scenario remains unchanged at lower EOc fractions. In the lower part of Fig. 4 the results are plotted for sample P(BO/EOc)38k-OH where the co-monomer fraction was reduced to 1.6 mol% and the calculated overall monomer to initiator ratio was 630. In this experiment the data points scatter more on a relative scale because of the very low vinly concentrations. The solid line represents the gravimetrically determined co-monomer fraction of the initial reaction mixture for the polymerization. Within the series of 1,2-epoxy-ω-alkenes their reactivities agree with their saturated counterparts, where it is known that the reactivity decreases with increasing alkyl chain length. However it was not expected that EH is more reactive than BO and EOc has the same reactivity as BO. A random copolymerization behavior was also found for the system ethylene oxide (EO) with differently functionalized glycidyl ethers.25 In that case similar reactivities of EO and the glycidyl ether derivatives can be explained on the basis of the electronic and steric properties of the co-monomers. In our case we do not yet have an explanation how the vinyl group could influence the reactivity of the monomer. The molecular weight characterization of sample P(BO/EOc)38k-OH is given in Table 1. From the 1H-NMR data the number of vinyl groups per chain was calculated to be 8.3 (Fig. 5).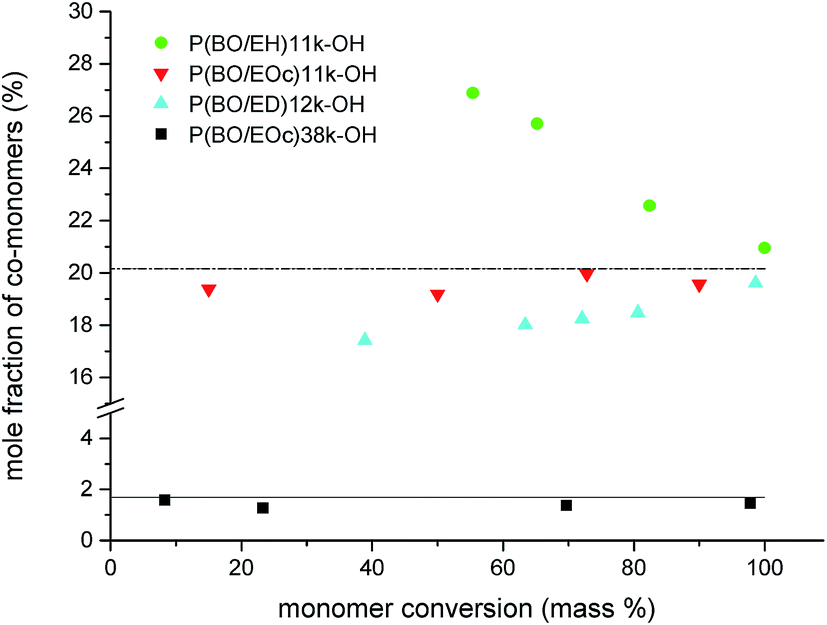 | ||
| Fig. 4 Co-monomer content of polymers during polymerization reactions of BO with different 1,2-epoxy-ω-alkenes. | ||
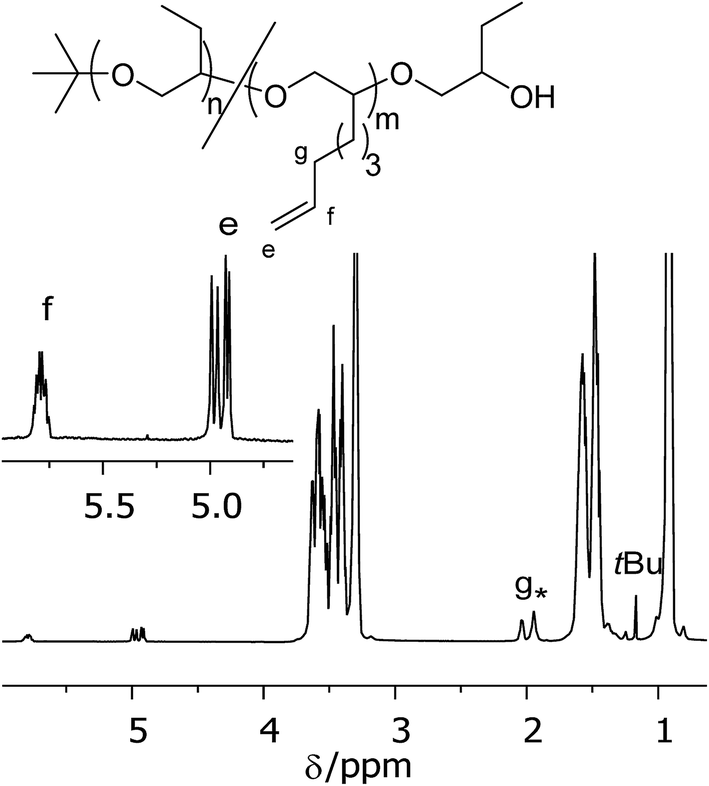 | ||
| Fig. 5 1H-NMR spectrum in CDCl3 of the fully polymerized sample P(BO/EOc)38k-OH. The insert shows the expanded vinyl region. The signal marked with * belongs to water. | ||
In the following reaction step vinyl groups of P(BO/EOc)38k-OH were first hydroborated with 9-BBN, followed by oxidation with H2O2/NaOH (Scheme 2). This reaction selectively generates terminal alcohol groups at the ends of the co-monomer side chains.17 The close inspection of the 1H-NMR spectrum of the oxidation product PBO38k(OH)8(OH) showed that no more than 1% of the original vinyl groups remained after the oxidation procedure (see ESI†). Fig. 6 shows the SEC chromatograms of P(BO/EOc)38k(OH) and PBO38k(OH)8(OH). They are identical except the small additional signal at about the double molecular weight in the trace of PBO38k(OH)8(OH). This signal covers about 1% of the overall signal intensity. In literature the coupling of chains via borate esters is described as a side reaction of this process.17a However, the high molecular weight material could not be removed under basic conditions where usually borate esters are cleaved. Therefore we assume a radically induced coupling process originating from H2O2. In this experiment a H2O2 excess of about 3 mol% was used and the reaction temperature did not exceed room temperature. The molecular weight characterization of P(BO/EOc)38k(OH) and PBO38k(OH)8(OH) is given in Table 1. Although the molecular weights are fairly high the values obtained from the NMR method coincide well with the SEC/LS results.
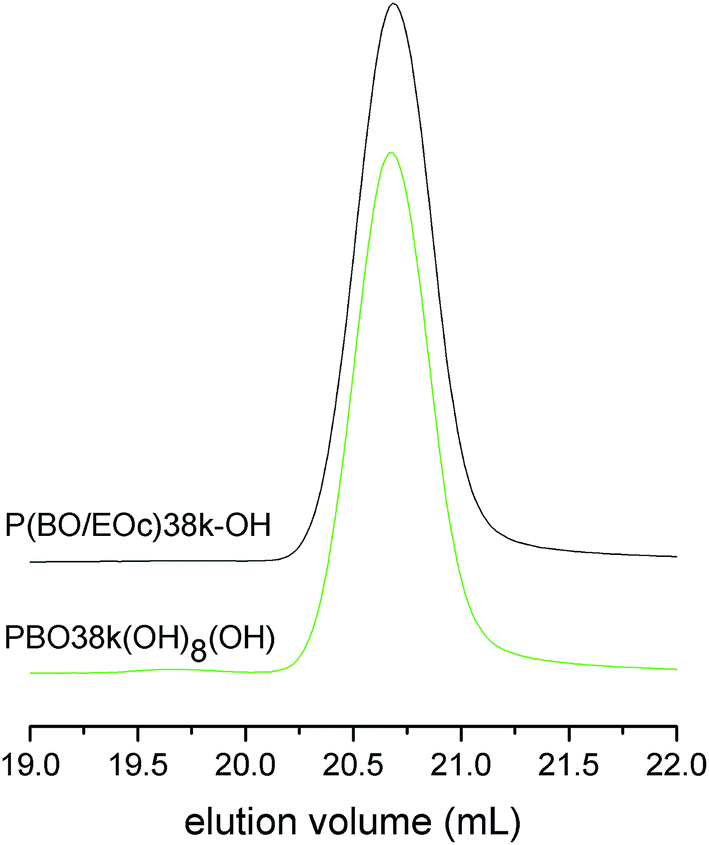 | ||
| Fig. 6 SEC chromatograms of P(BO/EOc)38k-OH and PBO38k(OH)8(OH). | ||
The alcohol groups of PBO38k(OH)8(OH) were converted into amino groups as described for the other polymers. The parent compound P(BO/EOc)38k-OH was not terminated with CH3I in order to block the chain end but synthesized with an OH-end-group. As a result of the elevated molecular weight the concentrations of chain ends are very small. Nonetheless the end-group signals are unambiguously visible in the 1H-NMR spectra and can be evaluated at least semi-quantitatively. The comparison of the signals related to the OH- and the NH2-end-groups (lower spectrum in Fig. 7, signals b and Y) shows that only about half of the end-groups in PBO38k(NH2)8(NH2) are functionalized with amino groups. This is understandable as the concentration of end-groups in the functionalization reactions was smaller by a factor of 8 compared to the mono- and difunctional variants. In addition the reaction time was reduced as the emphasis of these syntheses was the backbone functionalization.
 | ||
| Fig. 7 1H-NMR spectrum in pyridine-d5 of the functionalization sequence for PBO38k(OH)8(OH). | ||
Thymine and DAT functionalized polymers
Thymine functionalized polymers were synthesized using both alcohol and amino functional polymers (see Scheme 3). First thymine-1-acetic acid was coupled to the OH-functionalized polymers. TBTU was used to activate the thymine–acetic acid. The solvent was changed from the commonly used DMF to DMF/THF mixtures in order to dissolve the polymers. In contrast to the described conversion of low molecular weight molecules,26 almost no reaction was observed with the secondary alcohol groups of PBO5k1-OH. The backbone functionalized polymer PBO38k(OH)8(OH) reacted much better with about 7 out of 8 side chain OH-groups which were esterified while the terminal OH-groups remained at least largely unreacted (Fig. 8). Therefore no further attempts were made change the reaction conditions in order to improve the reactivity of the secondary chain ends.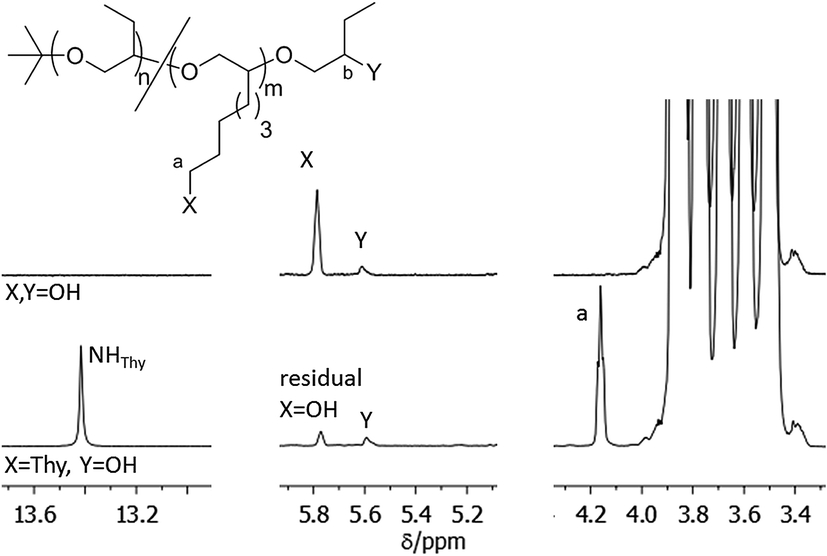 | ||
| Fig. 8 1H-NMR spectrum in pyridine-d5 of the thymine functionalization of PBO38k(OH)8OH by direct esterification. | ||
As a consequence, thymine end functionalized polymers PBO5k-Thy and Thy-PBO5k-Thy were synthesized from PBO5k-NH2 and H2N-PBO5k-NH2. The reaction conditions were similar to the previous esterification reactions except that the reaction time was doubled and the temperature was increased from room temperature to 40 °C. The functionalization reactions with DAT-Cl were carried out using the amino precursor in DMA in the presence of NaHCO3 following a modified literature procedure for the functionalization of bis-amino polypropylene oxide.9b,c The solvent had to be changed from EtOH/H2O to DMA to dissolve the less polar PBO. All PBO based polymers are soluble in DMA at higher temperatures. The reaction of PBO38k(NH2)8(NH2) with DAT-Cl was therefore carried out for 4 days at 80 °C to ensure full solubility of the polymer. For PBO5k-NH2 and H2N-PBO5k-NH2 the temperature was raised to 100 °C at a reaction time between 7 and 10 days to maximize the conversion of the sterically hindered secondary amine end-groups. In the case of DAT-PBO5k-DAT the slower conversion of the secondary versus the primary amine end-group could be observed from NMR samples that were taken to monitor the reaction progress. While the signal of the terminal methylene group of the primary amines (Fig. 9, signal a) vanishes in the DAT functionalized product, the methine proton of the secondary amine is still visible (Fig. 9, signal b). After the removal of the solvent under high vacuum conditions the polymers were solubilized in pentane to remove largely the excess of non-soluble DAT-Cl. Residual DAT-Cl also can be detected in the NMR spectra between 8.2 and 8.5 ppm. In the DAT functionalized polymers only traces of DAT-Cl could be detected which represented about 1% of the overall DAT content (Fig. 9). This compound shows a reduced solubility in any solvent and is difficult to remove using the usual precipitation work-up. Therefore, after the reaction and the solvent removal under high vacuum conditions the product mixture was solubilized in pentane and non-soluble DAT-Cl was separated by centrifugation.
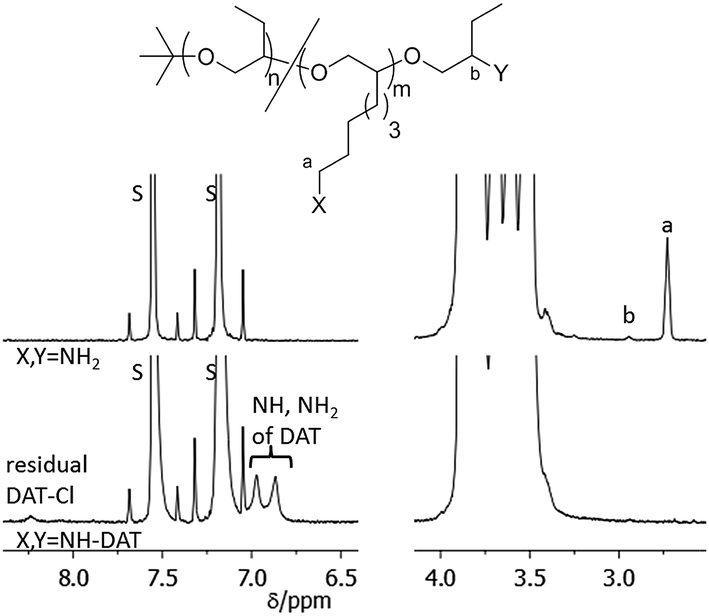 | ||
| Fig. 9 1H-NMR spectrum in pyridine-d5 of the DAT functionalization of PBO38k(NH2)8(NH2). The signals marked with S belong to residual pyridine protons. | ||
The SEC traces of all azide functionalized polymers coincide well with their alcohol precursors. Chromatograms of the amine functionalized polymers show very broad signals, presumably because of attractive interactions with the column material. The SEC results of the thymine functionalized polymers are given in Fig. 10. All polymers have narrow molecular weight distributions but contain small quantities of product having about double molecular weight. The measured values are listed in Table 1. Most of the high molecular weight fractions were present already in the alcohol functionalized precursors. Amine functionalized polymers are not added to Table 1 because of the artificial nature of the chromatograms. As this effect does not occur for the other polymers it is clear that the signal broadening is not a result of polymer decomposition.
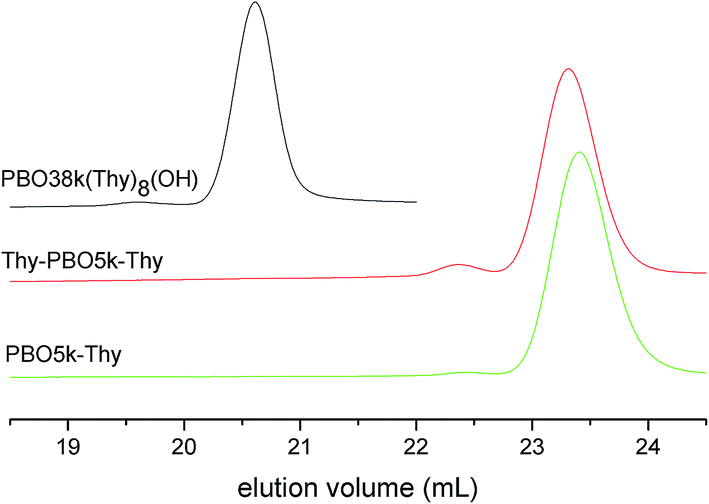 | ||
| Fig. 10 SEC chromatograms of PBO38k(O-Thy)8(OH), PBO5k-Thy and Thy-PBO5k-Thy. | ||
The scenario is different for the DAT functionalized polymers. All materials contain visible amounts of high molecular weight products (Fig. 11). The values extracted from the SEC curves are listed in Table 1. The higher the DAT content is the larger is the fraction of coupled chains. PBO38k(DAT)8(DAT) contains even a smaller fraction of product where more than two chains are coupled. To our knowledge the coupled chains are not a result of hydrogen bond formation between DAT groups of different polymers. The addition of thymine in the SEC experiment did not change the chromatograms. So far we do not have an explanation for the chain coupling reaction which occurs during the reaction with DAT-Cl. The observation that the amount of chain coupling corresponds to the number of functionalization sites suggests involvement of the DAT groups in the coupling process. The commercial 2-chloro-4,6-diamino-1,3,5-triazine that was used in the functionalization has a purity of ∼98%. While polychlorinated impurities could explain the coupling reactions, we can exclude the presence of 2,4-dichloro-6-amino-1,3,5-triazine in DAT-Cl. The absence of 2,4-dichloro-6-amino-1,3,5-triazine was proved in a separate NMR analysis of DAT-Cl. The increased temperature and long reaction time do not seem to contribute to the coupling process as we were able to detect similar dimerization in the DAT functionalization of diamino-polypropyleneoxide which proceeds at lower temperature and shorter times.9b,c
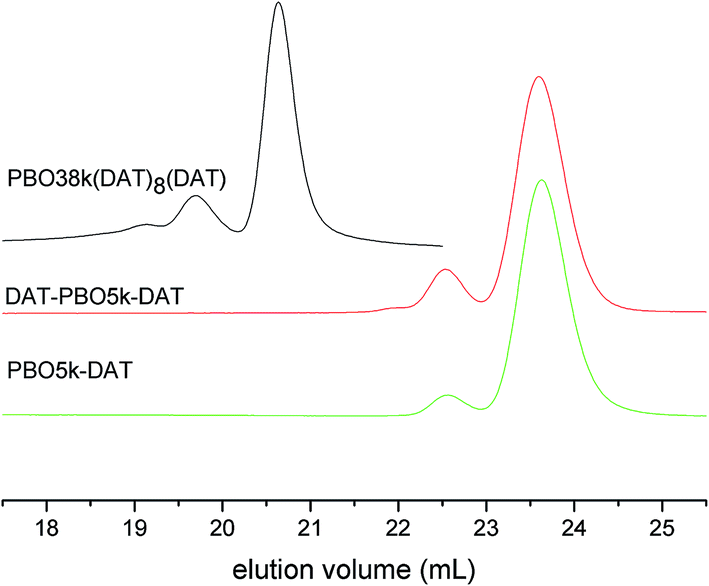 | ||
| Fig. 11 SEC chromatograms of PBO38k(DAT)8(DAT), PBO5k-DAT and DAT-PBO5k-DAT. | ||
Quantification of functionalization degrees
Thymine functionalized polymers can be obtained directly from the alcohol precursors. This procedure works only for primary alcohols. For the secondary chain ends the more reactive amine groups are required. DAT functionalized polymers are accessible only via the amine precursors. High functionalization degrees require elevated reaction temperatures in combination with long reaction times.The functionalization degrees with DAT and thymine groups can be calculated from the final product spectra by comparing the 1H-NMR signals related to these groups with the end-group signals of residual precursors. In addition absolute functionalization degrees were calculated from the comparison of the DAT or thymine functional group signals with the signal of the tert-butyl initiator unit. For the monofunctional polymers the values are consistent with the relative values (Table 2). This shows the absence of other so far disregarded chain end functionalities. For the bifunctional polymers only relative functionalization degrees can be obtained. The almost quantitative functionalization of the bifunctional variants with DAT and thymine is mainly a result of the optimized synthesis of the amine precursors. For the backbone functionalized polymers only PBO38k(Thy)8(OH) could be used to calculate absolute functionalization degrees. From the absolute thymine functionality and considering the residual alcohol content an overall number of 8.5 side chain functional groups per chain was calculated. This is in very good agreement with the 8.3 vinyl groups per chain measured for the parent compound P(BO/EOc)38k-OH. The chain end functionalization of PBO38k(Thy)8(OH) and PBO38k(DAT)8(DAT) occurred rather poor. In the case of PBO38k(Thy)8(OH) this reaction was not optimized and for PBO38k(DAT)8(DAT) the method used does not allow better results under these extreme dilution conditions.
| Relative functionalization degrees from direct comparison of functional group related signals | Absolute functionalization degrees from comparison with tert-butyl unitb | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Primary chain ends | Secondary chain ends | ||||||||
| OH | N3 | NH2 | Thy/DAT | OH | N3 | NH2 | Thy/DAT | ||
| a nd: no measurable NMR signals exist in the case of secondary N3 functionalized polymers; full conversion was assumed on the basis of the IR measurements. b Only DAT or thymine fuctionalization. c Synthesized from the alcohol precursor. d Synthesized from the amine precursor. | |||||||||
| PBO5k-Thy | 0.03 | nd | 0.00 | 0.97 | 0.96 | ||||
| PBO5k-DAT | 0.05 | nd | 0.05 | 0.90 | 0.87 | ||||
| Thy-PBO5k-Thy | 0.00 | 0.00 | 0.00 | 1.00 | 0.00 | nd | 0.00 | 1.00 | |
| DAT-PBO5k-DAT | 0.01 | 0.00 | 0.00 | 0.99 | 0.02 | nd | 0.00 | 0.98 | |
| PBO38k(Thy)8(OH)c | 0.15 | 0.85 | ∼1 | 7.25 | |||||
| PBO38k(DAT)8(DAT)d | 0.02 | 0.00 | 0.02 | 0.96 | ∼0.6 | nd | ∼0.2 | ∼0.2 | |
Rheological characterization
Hydrogen bond forming groups are polar and tend to form not only directed interactions via the hydrogen bonds but also cluster. This is the case especially in non-polar environments. In this work we used the semi-polar PBO in order to suppress clustering effects. Linear rheology experiments were carried out with the backbone-functionalized sample PBO38k(Thy)8(OH) and the non-functional mother compound P(BO/EOc)38k-OH. Fig. 12a summarizes the storage and loss moduli, G′(ω) and G′′(ω) for both polymers versus the frequency ω, reduced to T0 = −25 °C. The data exhibit simple polymeric viscoelastic behavior i.e. a ω1 dependence of G′′(ω) at low frequencies, characteristic of viscous flow. Likewise ω2 is found for the elastic contribution. The sufficiently-well pronounced plateau region in G′(ω) in the intermediate frequency range is indicative of chain entanglements. The functionalization with the thymine groups slightly slows down the dynamics due to an enhanced friction of the larger side groups and a rise in the glass transition temperature by ∼4 °C. A non-specific clustering as observed in literature can be fully ruled out. Within statistical uncertainty the plateau modulus of ∼0.29 MPa is unaffected by the functionalization and corresponds to an average mass between entanglements (Me) of about 8.000 g mol−1.16 The effect of the DAT functionalization was analyzed by comparing the viscoelastic behavior of the functionalized compound PBO5k-DAT and the non-functionalized precursor PBO5k1-OH. In Fig. 12bG′(ω) and G′′(ω) for both polymers versus ω are reported reduced to T0 = −35 °C. The DAT groups affect the dynamics of the short chains, which in their parent form show a simple Rouse behavior typical of a linear polymer with a molecular weight below the critical molecular weight Mc = 2Me. The DAT derivative has a Tg ∼ 3 °C higher than the non-functionalized mother compound. It shows a shallow shoulder in the intermediate frequency range which can be attributed to the association of the DAT groups. The Rouse model was used to fit the data for both compounds. The molecular weight found for PBO5k1-OH is in agreement with the values reported in Table 1. PBO5k-DAT can only be described as a mixture of ∼40% unimers and ∼60% dimers. From the results obtained, an association constant for the DAT groups on the order of 20 was estimated.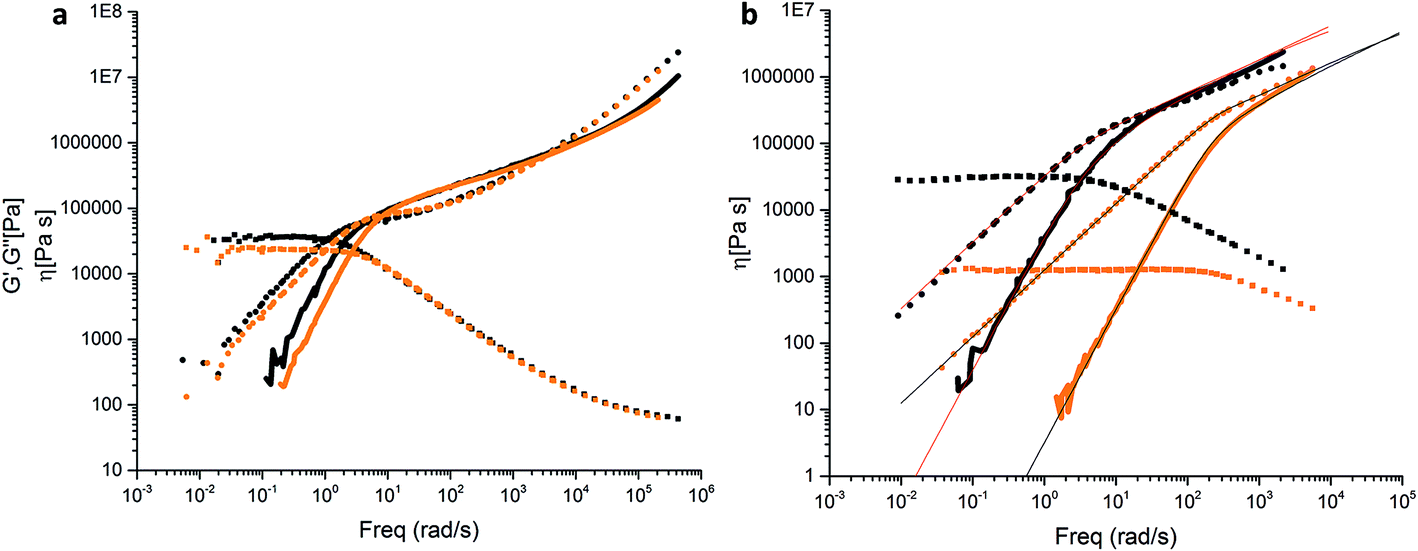 | ||
| Fig. 12 Storage moduli G′(ω) (solid lines), loss moduli G′′(ω) (dotted lines) and viscosities (squares) for (a) P(BO/EOc)38k-OH (orange) and PBO38k(Thy)8(OH) (black) and (b) PBO5k1-OH (orange) and PBO5k-DAT (black) including the Rouse model fit for PBO5k1-OH and PBO5k-DAT. | ||
In the next step a mixture of PBO38k(Thy)8(OH) and PBO5k-DAT was examined (Fig. 13a). The molar ratio of thymine to DAT groups was 1.0. In addition the respective mixture of PBO38k(Thy)8(OH) and PBO5k1-OH was studied. This blend shows a Rouse-like behavior which arises from the dilution of the tube of the entangled backbone with the shorter chains. These act as a solvent and decrease the entanglement density. The dilution effect is a function of the volume fraction of the short arms. The modulus of this system is related to the plateau modulus of the backbone chains as GN(ϕ) = GNϕα+1 where the dilution exponent α assumes values between 1 and 4/3. In this examination α = 1 was chosen because of the theta conditions. The Rouse-like behavior of the diluted backbone chains occurs when the diameter of the diluted tube equals the end-to-end distance27 of the backbone chains.
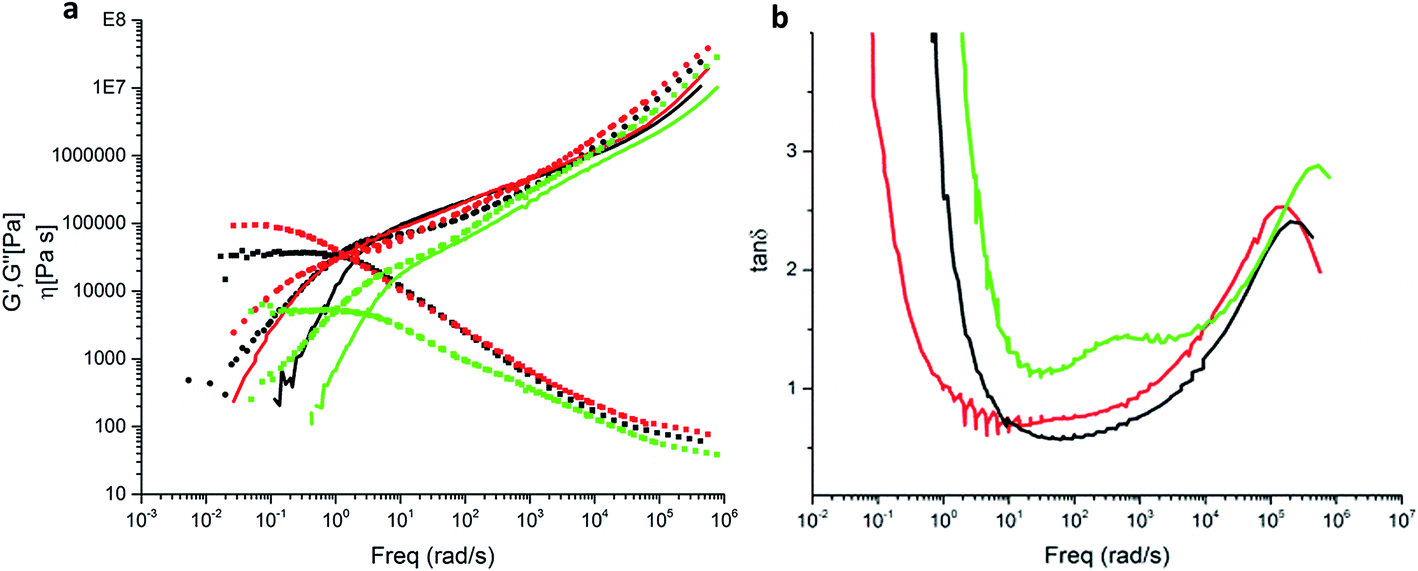 | ||
| Fig. 13 (a) Storage moduli G′(ω) (solid lines), loss moduli G′′(ω) (dotted lines) and viscosities (squares) for PBO38k(Thy)8(OH) (black), PBO38k(Thy)8(OH)/PBO-5k-DAT in a 1:1 molar ratio of Thy/DAT (red) and PBO38k(Thy)8(OH)/PBO5k1-OH in a 1:1 molar ratio of Thy/PBO5k1-OH (green) (b) respective tan of the phase angle. | ||
In the presence of the supramolecular groups the observed dynamics is different. In this case a plateau in the intermediate frequency region was observed. This plateau modulus is comparable to the one observed for the undiluted backbone. The observed plateau can be attributed exclusively to the association of the supramolecular groups. For the thymine–DAT association the formation of a transient comb polymer is expected. A covalent comb shows a pronounced hierarchical relaxation time spectrum. This is absent in our case due to the transient character of the bonds and low molecular weight of the arms which act merely as friction points in the polymer chain.
Fig. 13b shows the effect of the arms in blends with the backbone PBO38k(Thy)8(OH). In this case tanδ = G′′(ω)/G′(ω) was used, which is more sensitive in distinguishing different relaxation processes associated to different components in the blends. In the mixture with PBO5k1-OH the peak around 500 rad s−1 is attributed to the arm component. In the mixture with PBO5k-DAT a broader shoulder becomes visible around 10 rad s−1. It is to be noted that the strength of the latter peak is temperature-dependent as it corresponds to the relative fraction of bonded arms.
Conclusions
In this work we have shown the synthesis of 1,2-poly(butylene oxide) being functionalized at the chain ends or randomly at the backbone with thymine or DAT groups. The use of pyridine-d5 as solvent for the NMR analyses allowed monitoring the conversions by the decrease of signals corresponding to the starting material. This method is more precise than the frequently used comparison of end-group signals and polymer signals. In the latter case the functionalization degrees can only be calculated with the knowledge of the polymer molecular weight, which can only be determined with a precision of ±5 to 10%. Our NMR approach is especially useful for side-chain functionalized polymers, which have a variable number of functional groups per chain. In this case MALDI based methods fail in most of the cases because of too complex spectra. In addition NMR allows quantifying small fractions of different functionalities, which can be difficult using MALDI. Our results have shown that especially polymers being pre-functionalized with amino groups allow attaching thymine and DAT groups with high functionalization degrees and using commercially available supramolecular precursors. Even the less reactive secondary chain ends could be functionalized basically quantitatively in both cases, the pre-functionalization with amino groups and the subsequent functionalization with supramolecular groups. Initially a functionalization via click chemistry of the intermediate azide compounds was considered as an alternative to the amidation/esterification reactions described in this work. The synthesis of DAT and thymine terminated PIB via copper catalyzed alkyne–azide cycloaddition (CuAAC) has been described by Herbst et al.9a However, in our synthesis of thymine and DAT functionalized PBO the click conditions proved to be inferior compared to the classic amidation/esterification. All reagents for the amidation/esterification steps (2,4-diaminotriazinechloride and thymineacetic acid) are commercially available. In addition, a major disadvantage of the CuAAC reaction is the difficulty to purify the polymer and fully remove the copper catalyst. This problem is well known in CuAAC reactions and is probably enhanced in our case by the presence of thymine and DAT, which can potentially act as ligands for the copper impurities.28 The results obtained by linear rheology show a significant effect of the supramolecular interactions on the dynamics of the polymer system. These results evidence that thymine–DAT association leads to the formation of a transiently branched system and is dominant in the melt state. Although the hierarchical relaxation processes typical for a comb-like polymer are not observed, the formation of the transient comb is revealed by the differences observed in the dynamics of the supramolecular system and the inactive blend. In addition, the rheological data clearly show that neither the thymine nor the DAT groups exhibit non-directed interactions in the PBO melt. The molecular structure of the assembly is the topic of an upcoming work in which the supramolecular association is studied in terms of temperature and comb functionality.Acknowledgements
Zhang Wei is grateful to the support of Research Project Supported by Shanxi Scholarship Council of China (2013-151). The authors thank Viktoria Iflaender (Fachhochschule Aachen, Germany) for her synthesis support.References
- J.-M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 ( Angew. Chem. , 1988 , 100 , 91–116 ) CrossRef.
- J.-M. Lehn, Makromol. Chem., Macromol. Symp., 1993, 69, 1–17 CrossRef CAS.
- (a) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Shenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (b) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193–207 RSC.
- Self-Healing Polymers, From Principles to Applications, ed. W. H. Binder, Wiley-VCH, Weinheim, 2013 Search PubMed.
- M. Weck, Polym. Int., 2007, 56, 453–460 CrossRef CAS.
- (a) D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC; (b) R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed; (c) Hydrogen Bonded Polymers, Adv. Polym. Sci., ed. W. H. Binder, 2007, vol. 207 Search PubMed.
- (a) P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2006, 29 CrossRef CAS; (b) T. J. Mooibroek and P. Gamez, Inorg. Chim. Acta, 2007, 360, 381–404 CrossRef CAS.
- (a) F. Herbst, K. Schröter, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006–10016 CrossRef CAS; (b) J. Cortese, C. Soulié-Ziakovic, M. Cloitre, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2011, 133, 19672–19675 CrossRef CAS PubMed; (c) J. Cortese, C. Soulié-Ziakovic, S. Tencé-Girault and L. Leibler, J. Am. Chem. Soc., 2012, 134, 3671–3674 CrossRef CAS PubMed; (d) I. German, F. D'Agosto, C. Boisson, S. Tencé-Girault and C. Soulié-Ziakovic, Macromolecules, 2015, 48, 3257–3268 CrossRef CAS.
- (a) F. Ilhan, M. Gray and V. M. Rotello, Macromolecules, 2001, 34, 2597–2601 CrossRef CAS; (b) A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell and V. M. Rotello, Nature, 2000, 404, 746–748 CrossRef CAS PubMed; (c) B. L. Frankamp, O. Uzun, F. Ilhan, A. K. Boal and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 892–893 CrossRef CAS PubMed.
- K. Hackethal, F. Herbst and W. H. Binder, Polym. Chem., 2012, 50, 4494–4506 CrossRef CAS.
- A. R. Brás, C. H. Hövelmann, W. Antonius, J. Teixeira, A. Radulescu, J. Allgaier, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, Macromolecules, 2013, 46, 9446–9454 CrossRef.
- T. Yan, K. Schroeter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Macromolecules, 2014, 47, 2122–2130 CrossRef CAS.
- F. Herbst and W. H. Binder, Polym. Chem., 2013, 4, 3602–3609 RSC.
- S.-W. Kuo and H.-T. Tsai, Macromolecules, 2009, 42, 4701–4711 CrossRef CAS.
- C. Gerstl, G. J. Schneider, W. Pyckhout-Hintzen, J. Allgaier, D. Richter, A. Alegría and J. Colmenero, Macromolecules, 2010, 43, 4968–4977 CrossRef CAS.
- (a) U. Bayer and R. Stadler, Macromol. Chem. Phys., 1994, 195, 2709–2722 CrossRef CAS; (b) T. C. Chung, M. Raate, E. Berluche and D. N. Schulz, Macromolecules, 1988, 21, 1903–1907 CrossRef CAS.
- J. Allgaier, S. Willbold and T. Chang, Macromolecules, 2007, 40, 518–525 CrossRef CAS.
- D. A. Pearson, M. Blanchette, M. L. Baker and C. A. Guindon, Tetrahedron Lett., 1989, 30, 2739–2742 CrossRef CAS.
- A. C. French, A. L. Thompson and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 1248–1252 ( Angew. Chem. , 2009 , 121 , 1274–1278 ) CrossRef CAS PubMed.
- D. S. Noyce and J. A. Virgilid, J. Org. Chem., 1972, 37, 2643–2647 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, Protecting Groups in Organic Synthesis, Wiley VCH, Weinheim, 4th edn, 2006 Search PubMed.
- M. Ito, K. Koyakumaru, T. Ohta and H. Takaya, Synthesis, 1995, 376–378 CrossRef CAS.
- S. E. Grieshaber, A. J. E. Farran, S. Lin-Gibson, K. L. Kiick and X. Jia, Macromolecules, 2009, 42, 2532–2541 CrossRef CAS PubMed.
- (a) C. Mangold, B. Obermeier, F. Wurm and H. Frey, Macromol. Rapid Commun., 2011, 32, 1930–1934 CrossRef CAS PubMed; (b) B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 ( Angew. Chem. , 2011 , 123 , 8136–8146 ) CrossRef CAS PubMed; (c) C. Mangold, B. Obermeier and H. Frey, Polym. Chem., 2012, 3, 1714–1721 RSC.
- J. K. Twibanire and T. B. Grindley, Org. Lett., 2011, 13, 2988–2991 CrossRef CAS PubMed.
- C. Gerstl, G. J. Schneider, W. Pyckhout-Hintzen, J. Allgaier, S. Willbold, D. Hoffmann, U. Disko, H. Frielinghaus and D. Richter, Macromolecules, 2011, 44, 6077–6084 CrossRef CAS.
- N. Jasinski, A. Lauer, P. J. M. Stals, S. Behrens, S. Essig, A. Walther, A. S. Goldmann and C. Barner-Kowollik, ACS Macro Lett., 2015, 4, 298–301 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full 1H-NMR, 1H,1H COSY and 1H,13C HSQC spectra of the products and IR spectra of the azide as well as amine products. See DOI: 10.1039/c5ra24547h |
| ‡ Present address: Shanghai Fine Chemical Co. LTD., 201505 Shanghai, P. R. China. |
| This journal is © The Royal Society of Chemistry 2016 |