
One-pot synthesis of amides from carboxylic acids activated using thionyl chloride†
Abstract
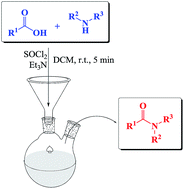
A one-pot synthesis of secondary and tertiary amides from carboxylic acids and amines by using SOCl2 has been developed. Also when sterically hindered amines were used as the starting materials, excellent yields of the corresponding amides were obtained. The amidation of N-protected α-amino acids with secondary amines proceeds effectively with good yields. The process works well also in the presence of acid sensitive groups and occurs with almost complete retention of stereochemical integrity of chiral substrates. This protocol could be extended to industrial large-scale production processes.
 Please wait while we load your content...
Please wait while we load your content...

