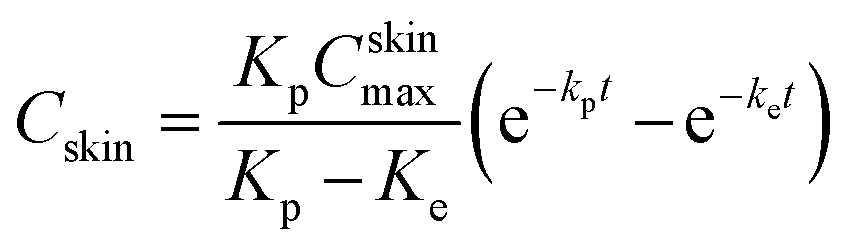DOI:
10.1039/C5RA24516H
(Paper)
RSC Adv., 2016,
6, 20713-20727
Aceclofenac–β-cyclodextrin-vesicles: a dual carrier approach for skin with enhanced stability, efficacy and dermatokinetic profile
Received
19th November 2015
, Accepted 14th February 2016
First published on 16th February 2016
Abstract
The aim of the current investigation was to develop and characterize lipid-based carriers of aceclofenac (ACE) with enhanced stability and transdermal delivery potential to the inflammatory sites in osteoarthritis. An attempt was made to complex the drug with a biocompatible complexing agent, i.e., β-cyclodextrin and the same was further encapsulated in the lipid bilayers of liposomes. FT-IR studies depicted the masking of one of the functional groups of ACE indicating the interaction of the drug–CD complex with the lipid bilayers of the prepared liposomes. The values of particle size, polydispersity index (PDI), zeta potential of the developed carrier system were found to be 481.7 nm, 0.214 and −29.54 mV, respectively. The system was further incorporated in a hydrogel which was found to be a shear-thinning system with a yield value of 3.625 Pa and viscosity of 3.085 Pa s. Skin permeation studies revealed the superiority of the prepared ACE loaded-β-CD liposomal gel over the MKT gel with an enhancement ratio of 2.69. Skin irritancy studies performed on LACA mice skin proved the safety and non-irritancy of the prepared formulation. The dermatokinetic studies confirmed better permeation and enhanced skin bioavailability of ACE to epidermis as well as dermis vis-à-vis the MKT product. The developed system not only improved the delivery aspects of ACE, but also offered substantial stability to this highly hydrolysis labile molecule. The current findings provide a lead for the development of an effective topical formulation of ACE with substantial stability in β-cyclodextrin-vesicles.
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most prescribed and “over-the-counter” used drugs for various non-neuronal pain-related diseases like arthritis,1,2 spondylitis and gout.3 Though widely preferred, the most common route, i.e., the oral route of administration of NSAIDs in chronic treatment of these disorders leads to serious consequences such as gastrointestinal ulceration and bleeding,4 cardiovascular side effects such as hypertension, congestive heart failure, renal failure, hepatotoxicity,5 idiosyncratic and skin reactions. Topical drug delivery, on the other hand, offers many advantages over the oral route including avoidance of hepatic metabolism, ease of administration, localized site of action and lack of systemic side effects. However, this route also poses challenges of poor delivery due to ineffective penetration of the drugs across different barrier layers of the skin and local irritation.6 These limitations of ineffective topical delivery are plausibly due to faulty formulation design employing conventional components and techniques.7 The new approaches employ biocompatible components and are emerging as panacea for various problematic drugs.8,9 Various nanocolloidal carriers have been prepared in the recent past and the targets of safe and effective topical delivery have been achieved.7 Aceclofenac (ACE), a derivative of diclofenac, approved by US FDA in 1992 is a selective potent inhibitor of COX-2, an inducible enzyme responsible for the generation of inflammatory mediators, and is in the target for the management of rheumatoid arthritis, osteoarthritis, ankylosing spondylitis and musculoskeletal pain.10,11 It is a weak acid (pKa = 4.7) practically insoluble in water and acidic environment, possesses poor aqueous solubility of the order of 60 μg mL−1, but is highly permeable according the Biopharmaceutical Classification System (BCS II).12 Aceclofenac has a plasma elimination half-life of approximately 4 hours.13 Due to its short half-life, it is mandatory to be administered frequently in order to maintain the desired concentrations.14 Therefore, a sustained release formulation via topical route can serve the dual purpose of enhancing the duration of action and elimination of the systemic side effects.7,15
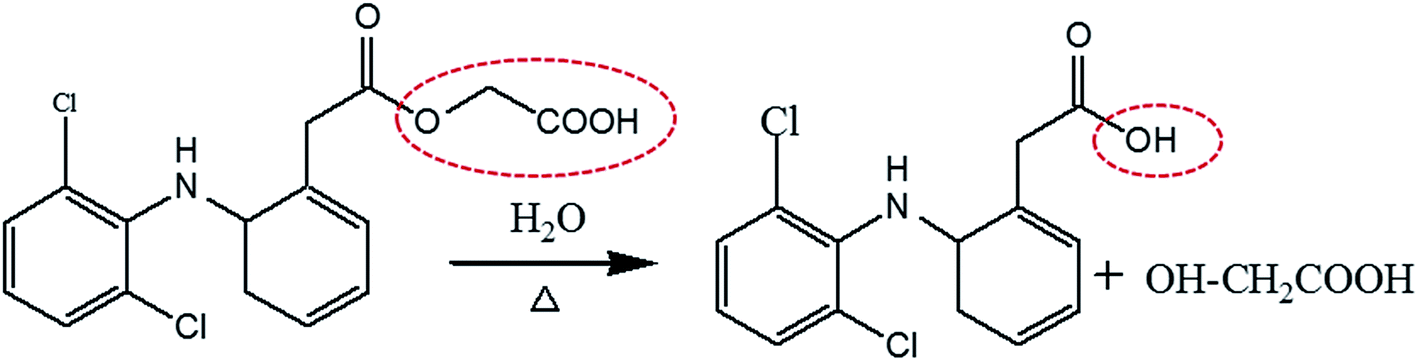
Besides poor penetration and delivery, the problem of degradation poses a colossal challenge for the pharmaceutical scientists to develop a stable formulation to meet the market needs.16 ACE is chemically unstable in acidic, alkaline and neutral media as well as prone to photodegradation. A significant degradation of ACE has been reported earlier in various literature reports.14,16 Being an ester of glycolic acid, ACE is highly prone to hydrolysis and degrades into diclofenac as shown in Fig. 1. The instability of ACE in aqueous environment not only decreases its efficacy and safety but also makes it difficult to be formulated. Therefore, the present work aims to develop a stable topical formulation of ACE with marked potential to deliver this poor penetrating molecule to various skin layers in safe and effective manner for the local treatment of pain-related disorders.
 |
| | Fig. 1 Ester linkage hydrolysis of ACE to give diclofenac and glycolic acid. | |
2. Materials
Aceclofenac (ACE; M/s Ipca Laboratories Pvt. Ltd., Mumbai, India) and phosphatidylcholine (PL; Phospholipon 90G; M/s Phospholipid GmbH, Nattermannallee, Germany) were obtained ex-gratis by the respective companies. Carbopol® 934 was obtained as a generous gift sample from M/s Panacea Biotec Ltd., Lalru, Punjab, India. Sephadex G-50 (M/s Sigma-Aldrich, St. Louis, USA), cholesterol (Chol; M/s Avanti Polar Lipids, Alabaster, USA), hydroxypropyl β-cyclodextrin (HP-β-CD) and β-cyclodextrin (β-CD) (M/s Sigma Chemical, St. Louis, USA) were procured from the respective sources. All other chemicals used were of analytical grade. Ultrapure water (Milli-Q® Integral system; M/s Merck Millipore, Billerica, USA) was employed throughout the study.
3. Methods
Considering the photostability issues with ACE, all the formulation and characterization studies were performed in amber colored glass wares in the dark.16
3.1. Preparation of ACE–β-cyclodextrin complexes
ACE–β-cyclodextrin complexes were prepared by the well reported slurry method.17,18 In brief, ACE and β-CD were complexed in the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, firstly adding β-CD in a glass mortar, followed by addition of water. Then after, weighed quantity of drug was added slowly by grinding, and the mixture was grounded for 1 h. The obtained solid mass was further dried under vacuum and pulverized.
:1, firstly adding β-CD in a glass mortar, followed by addition of water. Then after, weighed quantity of drug was added slowly by grinding, and the mixture was grounded for 1 h. The obtained solid mass was further dried under vacuum and pulverized.
3.2. Characterization of ACE–β-CD complex
3.2.1. Differential scanning calorimetry (DSC). The DSC thermograms were recorded with a DSC Q20 TA (M/s PerkinElmer Inc. USA) instrument by using standard aluminum pans. An empty pan was used as a reference pan. The samples (2–5 mg) were heated at a scanning rate of 10 °C min−1 over a temperature range between 20–350 °C.19
3.2.2. Fourier transform-infrared spectroscopy (FT-IR). Fourier transform infrared spectroscopy of (drug) as well as β-CD and Ace–β-CD complex was carried out by using FT-IR instrument (Spectrum Two™ M/s PerkinElmer Inc., USA) at 25 °C and wave number range from 3500 cm−1 to 500 cm−1. The samples were mixed thoroughly with potassium bromide in the mass ratio of 2:98.19
3.2.3. Phase solubility analysis. An excess amount of ACE (100 mg) was added to 25 mL of the aqueous solution (pH 7.0) containing various concentrations of β-CD (3–15 mM) and shaken at 30 °C for 24 h on a rotary flask shaker. After equilibration, the samples were withdrawn and filtered using 0.5 μm membrane filters. The samples were assayed employing RP-HPLC analysis and the apparent stability constant (Kc) was calculated.20
3.3. Preparation of ACE–β-CD liposomes
ACE–β-CD liposomes were prepared by the thin film hydration method.21–23 Accurately weighed quantities of phospholipid (6% w/w) and cholesterol (3% w/w) were dissolved in chloroform: methanol (1:1) in a round bottom flask. The solvent was evaporated at 55 ± 2 °C under reduced pressure (at 120 rpm) using rotary film evaporator (Buchi RE 121, Switzerland). After complete evaporation of the solvent, the obtained thin lipid film was hydrated at 35 ± 2 °C with aqueous phase (distilled water) containing accurately weighed quantity of ACE–β-CD complex using rotary film evaporator (at 50 rpm) to obtain a homogenous suspension of β-CD-liposomes. The suspension was kept for 24 h at room temperature for complete hydration process.
3.4. Incorporation of liposomal suspension in secondary vehicle
To incorporate the β-CD-liposomes in topically applicable vehicle, these were further gelled with 1% w/w of neutralized Carbopol® 934 hydrogel.
3.5. Characterization of the developed ACE–β-CD-liposomes
3.5.1. Micromeritics and zeta potential measurement. The vesicle size, polydispersity index (PDI) and zeta potential of ACE–β-CD-liposomes were determined by dynamic light scattering (DLS) using Delsa™ Nano (M/s Beckman Coulter, India Pvt. Ltd., Mumbai, India) installed at Institute of Microbial technology (IMTECH), Chandigarh.24 The mean value of three repeated measurements for each sample was reported as the final observation.Zeta potential was determined by measuring the electrophoretic mobility of the charged particles under an applied electric field from the Doppler shift of scattered light at 25 °C and the electric field strength was around 23.2 V cm−1. The average zeta potential values were directly obtained from the instrument software.9,24
3.5.2. Confocal laser scanning microscopy (CLSM). Coumarin-6 (3-(2-benzothiazolyl)-N,N-diethylumbelliferylamine, 3-(2-benzothiazolyl)-7-(diethylamino)coumarin) loaded β-CD liposomes were prepared by mixing coumarin-6 at a concentration of 0.15 μmol mL−1 with 6% PL and 3% Chol solution. Dye-loaded β-CD liposomes without drug were applied to the rat skin for 6 h followed by rinsing the skin with PBS pH 7.4 and the fixed slides were prepared.25–27 Localization of fluorescence in the rat skin layers after experimentation was examined using a confocal laser scanning microscope (Olympus FV10i, M/s Olympus Singapore Pvt. Ltd., Singapore).
3.5.3. Scanning electron microscopy (SEM). The surface morphology of ACE–β-CD liposomal suspension was examined by a JSM-6100 scanning electron microscope (Japan Electron Optics, Tokyo, Japan) installed at Central Instrumentation Lab (CIL), Panjab University, Chandigarh. One drop of the vesicular suspension was spread homogeneously onto a glass slide and left to dry for 30 min. The samples were coated with thin layer of gold using a JFC-1100 ion sputter coater under 0.5 mbar argon atmosphere. Pictures were taken at an excitation voltage of 20 kV.28,29
3.5.4. Vesicle count. The number of vesicles per cubic mm were counted microscopically employing a haemocytometer. The vesicles in 80 small squares were counted and their number density was calculated using the following formula as shown in eqn (1).28| |
 | (1) |
3.5.5. pH measurement. To determine the pH of the ACE–β-CD-liposomes, the sample was diluted with distilled water in the ratio of 1:1 and pH values were noted employing glass electrode pH meter (Cyberscan 510, M/s Eutech Instruments Pvt. Ltd., Singapore).
3.5.6. Fourier transform-infra red spectroscopy (FT-IR). FT-IR spectroscopy of the solid samples, i.e., drug, PL as well as the lyophilized ACE–β-CD liposomal suspension was carried out using FT-IR instrument (Spectrum Two™ M/s PerkinElmer Inc., USA) at 25 °C and wave number range from 3500 cm−1 to 500 cm−1. The samples were mixed thoroughly with potassium bromide in the mass ratio of 2:98 to form a pellet.19,30
3.5.7. Rheological studies. Rheological characterization of ACE–β-CD liposomal gel and marketed gel (MKT gel) was performed using a rotational type rheometer (Rheolab QC, M/s Anton Paar GmbH, Vienna, Austria) attached with a water jacket (C-LTD80/QC) for maintaining constant temperature. Data analysis was carried out using in-built instrument software Rheoplus/32, version 3.40. A DG26 spindle geometry was used for measuring the torque as well as viscosity of the sample. The optimized formulation was subjected to different shear stress and shear rate conditions and the measurements were carried out at room temperature. Temperature was kept at 30 °C and shear rate was selected from 0 to 100 s−1.15 All the tests were performed in triplicate and mean values were used in analysis. Power-law (eqn (2)) and Herschel–Bulkey model (eqn (3)) were employed to analyze the data of stress (τ) and shear rate (γ) as per the following equations.31where, k is consistency index; τ0 is yield stress and n is power-law exponent.
3.5.8. Texture analysis (spreadability). Texture profile analysis was performed to study other rheological characteristics, i.e., firmness and stickiness, of the formulations using TTC spreadability rig fitted on TA-XT2 Texture Analyzer™ (M/s Stable Micro Systems Ltd., UK).31
3.6. Ex vivo evaluation of the developed formulations: a comparative study
3.6.1. Animal ethical compliance. All the animal investigations were performed as per the guidelines of the Panjab University Animal Ethics Committee, duly approved for the purpose of control and supervision of experiments on animals by the Government of India with Ref. letter no. CAH/90H.
3.6.2. Ex vivo drug permeation studies. The ex vivo drug permeation studies were conducted on excised dorsal skin of Wistar rats on a two-compartment Franz diffusion cell assembly (PermeGear, Inc., Hellertown, PA, USA) equilibrated at 35 °C using thermostatically controlled water bath. The excised skin was mounted between the donor and receptor compartment exposing an effective area of 3.14 cm2 with sink volume of 30.0 mL. The system was stirred continuously to maintain the ideal sink conditions. The various developed formulations viz., ACE–β-CD liposomal gel, ethanolic drug solution and MKT gel formulation, each containing ACE equivalent to 1.5% w/w, were applied onto the mice skin in the donor compartment. Aliquots of 1 mL each were periodically withdrawn at suitable time intervals from the sampling port and replaced with an equal volume of fresh diffusion medium to maintain the constant receptor volume. The samples were assayed employing RP-HPLC analysis.24,27
3.6.3. Drug retention studies in skin. After completion of the permeation studies, the skin samples mounted on Franz diffusion cells were removed carefully and washed thrice, to remove any adhered formulation. The skin was then cut into small pieces and mixed with 5 mL ethanol and shaken for 24 h at 32 ± 1 °C for extraction of ACE.24,27 Supernatant was filtered through a membrane (0.22 μm). Subsequently, the filtrate was analyzed by HPLC at λmax 275 nm for drug concentration after suitable dilution.
3.7. In vitro drug deposition studies in dermal layers: dermatokinetic modelling
The whole skin was removed from the Franz diffusion cell at the respective sampling time intervals. The skin was washed thrice to remove any adhering formulation and subsequently soaked in hot water (60 °C) for the detachment of epidermis from dermis.24,25 Both the layers were then cut in to small fragments and mixed with 10 mL ethanol and shaken for 12 h at 32 ± 1 °C for complete extraction of ACE. Supernatant was filtered through a 0.22 μm membrane filter. The filtrate was analyzed for the drug content by the HPLC. The obtained data were fitted into one-compartment open model, as per the following (eqn (4)).32| |
 | (4) |
where Cskin is the concentration of drug in skin at time t, Kp is the dermal permeation constant, Cskinmax is the maximum concentration achieved in skin, and Ke is the skin elimination constant. Win-Nonlin Ver 5.0 software was employed to compute various dermatokinetic parameters, namely, Kp, Cskinmax, Ke, and Tskinmax (time required to achieve Cskinmax) and area under the curve (AUC0–12h) using the Wagner–Nelson method.33
3.8. Stability studies
The prepared ACE–β-CD liposomal gel formulation(s) were subjected to stability studies at 5 ± 3 °C and 40 ± 2 °C/75 ± 5% RH as per ICH guide lines, Q1A (R2) for 3 months in order to determine their shelf-life and storage condition (ICH, 1996).34
3.9. Skin compliance studies
For skin compliance studies, modified Draize patch test was used27,35 to evaluate the skin compliant nature of the prepared ACE–β-CD liposomal gel formulation vis-à-vis the MKT gel formulation. Female Laca mice (3–5 weeks old; 20–25 g), were divided into 3 groups, consisting of 4 animals each. Group A received normal saline (i.e., untreated), Group B received MKT gel formulation (1.5%, w/w ACE) and Group C received ACE–β-CD liposomal gel formulation (1.5%, w/w ACE). The hair on the dorsal side of mice were removed with the help of a 0.1 mm animal hair clipper, 24 h prior to the application of the formulations. Formulations, 0.5 g (equivalent to 1.5% ACE), were applied on the hair free skin of mice by uniform spreading within the area of 4 cm2 once-a-day treatment for 7 days. Prior to each daily application, the formulation remaining on skin was removed carefully and swabbed with saline soaked cotton and the skin was observed for any visible change such as erythema. The mean erythemal scores were recorded (ranging from 0 to 4) depending on the degree of erythema; score 0 – no erythema; score 1 – slight erythema (light pink); score 2 – moderate erythema (dark pink); score 3 – moderate to severe erythema (light red) and score 4 – severe erythema (dark red). Animals were sacrificed by cervical dislocation and the skin samples were harvested and fixed in 10% buffered formalin solution. The sections were stained with hematoxylin and eosin and observed microscopically for histopathological attributes. The results were compared with the skin sections of untreated mice.27
4. Anti-inflammatory and analgesic activity
4.1. Carrageenan-induced paw oedema in rats
The anti-inflammatory activity of ACE–β-CD-liposomes was assessed by the well-established carrageenan-induced rat paw oedema model.36 The model was carried out on Wistar male rats. The animals were divided into four groups of 4 animals (n = 4) each. Group A was treated with plain β-CD liposomal gel formulation (control; formulation without drug), Group B was treated with the MKT gel formulation and Group C with ACE–β-CD liposomal gel formulation.
Animals were injected intra-plantar injection of 0.1 mL of 1.0% carrageenan solution in saline into the left hind paws of rats of all groups for local inflammation.37 Immediately after the administration of carrageenan, the paw volume was measured at time zero. The hind paw was immersed in the measurement cell up to the hair line to the ankle to determine the immersed paw volume in mL. This was measured by means of a volume displacement method using a plethysmometer (M/s. UgoBasile, Italy).38 After 30 min, different formulations were applied onto the surface of right hind paws by gentle rubbing. The paw volume was measured at 1, 2, 4, 6, 8, 10, 12 and 24 h time interval after carrageenan administration. The percentage paw volume increase from time zero was calculated. The time-course of the anti-inflammatory response was determined. The percent swelling of the paw was determined using following eqn (5).
| | |
Percent swelling (PS) = (V − V0) × 100
| (5) |
where,
V is the paw volume at different time intervals after the carrageenan injection and
V0 is the initial paw volume at time zero.
Further, the average percent paw swelling in the formulation treated group (i.e., Group B and C) was compared with control Group A and percent inhibition of the oedema formation was determined using following eqn (6).39
| | |
Percent inhibition of oedema = (1 − PSt/PSc) × 100
| (6) |
where, PSt is the percent swelling in formulation group and PSc is the percent swelling in control group.
39
4.2. Radiant heat tail-flick method
Analgesic effect of the prepared ACE–β-CD-liposomes was assessed by radiant heat tail-flick method employing an analgesiometer M/s. Adarsh Scientific, Ambala Cantt, India.31,36 The study was carried out in three groups (Group A, Group B and Group C) of LACA male mice of six animals each. The basal reaction time was measured for each group with cut-off period of 5 seconds. Group A animals were treated with β-CD liposomal gel formulation (control), Group B with MKT gel and Group C with ACE–β-CD liposomal gel formulation.
The reaction time was measured at desired time intervals for each group. The % analgesic effect was determined employing eqn (7) and One Way ANOVA was applied to the obtained data to decide on the difference in the values obtained.
| | |
Percent analgesic effect = (Tt − T0)/(T − T0) × 100
| (7) |
where,
T is cut off time period,
T0 is basal reaction time and
Tt is observed reaction time.
4.3. Statistical analysis
The results were statistically analyzed using one-way ANOVA followed by post hoc analysis using Student's t test. Statistical significance was considered at p < 0.05.
5. Results and discussion
5.1. Characterization of the ACE–β-CD-complex
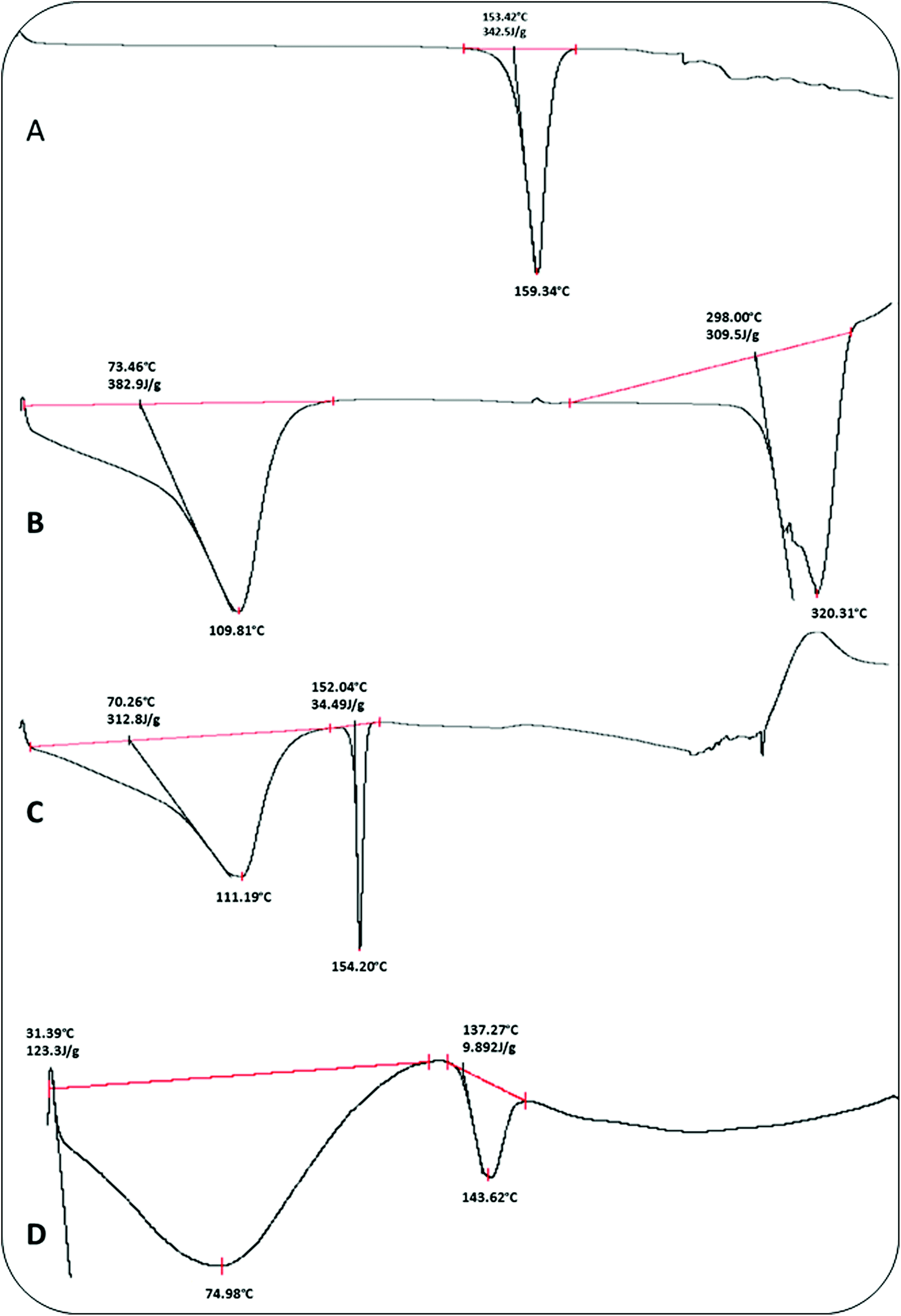
5.1.1. Differential scanning calorimetry (DSC). Fig. 2 depicts the DSC curves of ACE, β-CD, and the ACE–β-cyclodextrin complex (1:1). The DSC curve of aceclofenac showed an endothermal peak with an onset temperature at about 153 °C, which is mainly attributed to the melting of aceclofenac (149–153 °C).40 The DSC curve of the complex mainly showed the effect of β-CD, in which the characteristic endothermal peaks of aceclofenac disappeared. It can be considered that aceclofenac had been completely incorporated in β-CD and they had some interactions, such as the combination of hydrogen bonds and/or van der Waals force.41
 |
| | Fig. 2 DSC thermograms of (A) ACE, (B) β-cyclodextrin, (C) physical mixture (1 : 1) and (D) ACE-β-cyclodextrin complex (1 : 1). | |
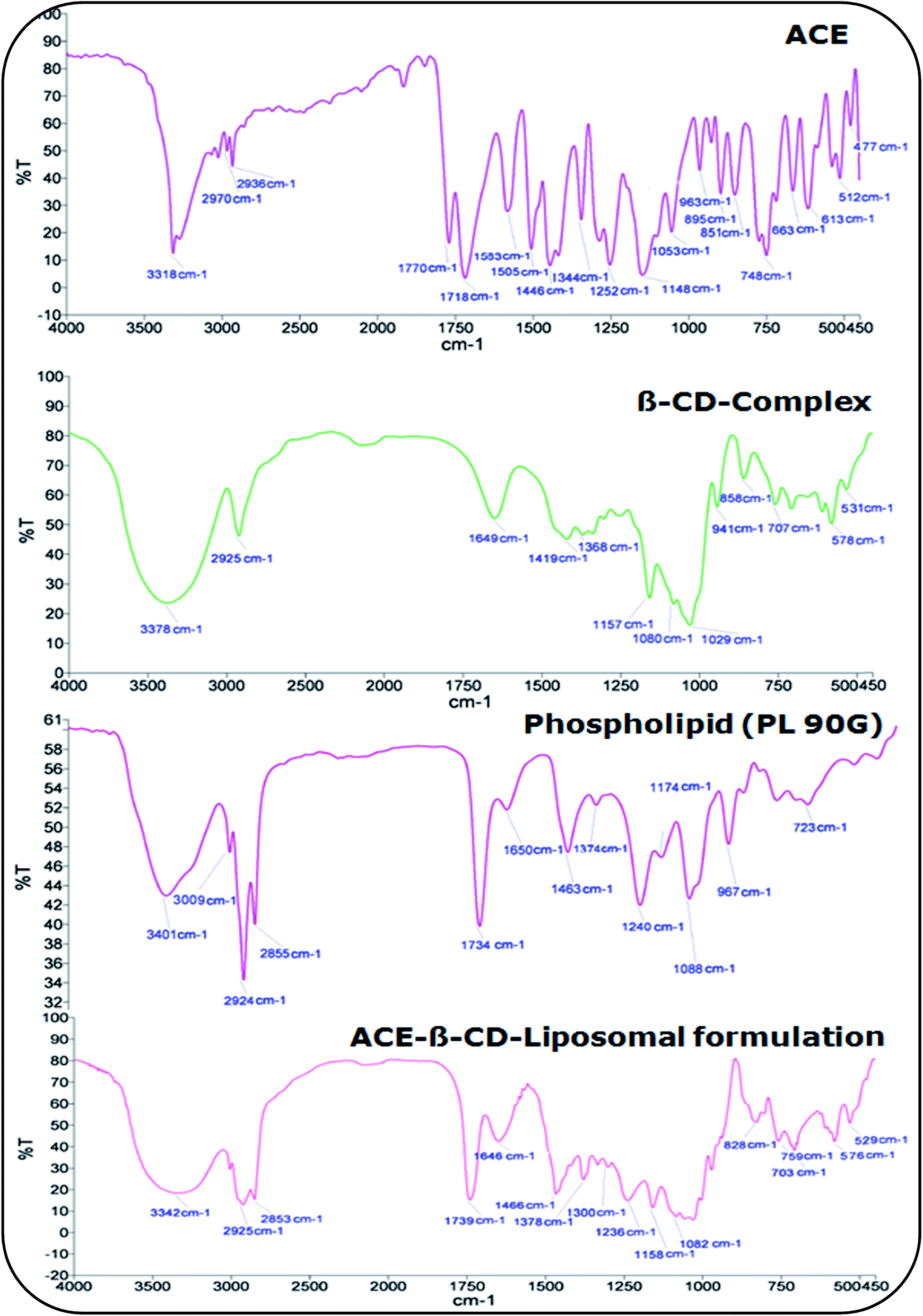
5.1.2. Fourier transform-infrared spectroscopy (FT-IR). The use of FT-IR technique allowed the detection of complex formations in solid phase by analyzing the significant changes in the shape and position of the absorbance bands of ACE, β-CD and inclusion complexes. Fig. 3 illustrates the IR peak of the solid ACE, β-CD and inclusion complex. The β-CD exhibited significant peaks 3378 cm−1, 2925 cm−1 and 1649 cm−1.42 While, ACE showed FTIR peaks at wave number of 3318 cm−1, 1770 cm−1, 583 cm−1 1505 cm−1, 1252 cm−1 and 1053 cm−1 which confirms the purity of drug sample as per the established standards.43 While ACE-β-CD complex had maximum absorption peaks at 3319 cm−1, 2927 cm−1, 1770 cm−1, and 1717 cm−1. In ACE–β-CD complex, the peaks of ACE were masked or shifted due to the intermolecular interactions. All the IR spectra of drug with excipients showed the above-mentioned characteristic peaks with insignificant shift and reduced intensity. This confirms the formation of complex.
 |
| | Fig. 3 IR-spectra of ACE, β-CD-complex and ACE–β-CD-complex. | |
5.1.3. Phase solubility analysis. The complexation of ACE with β-cyclodextrin was investigated by the phase solubility analysis. The phase solubility diagram for complex formation is depicted in Fig. 4. The aqueous solubility of ACE increased linearly as a function of concentration of CD. The enhancement in solubility observed is attributed to the formation of a 1:1 M inclusion complex.44
 |
| | Fig. 4 The phase solubility diagram of ACE–β-CD complex (1:1). | |
5.2. Characterization of the developed ACE–β-CD-liposomes
5.2.1. Micromeritics and zeta potential measurement. The average vesicle size of ACE–β-CD-liposomes was found to be 481.7 nm, as depicted in Fig. 5. The PDI value of the developed system was found to be 0.214, assuring narrow distribution of the polydispersed phase.24
 |
| | Fig. 5 Vesicle size distribution and PDI of ACE–β-CD-liposomes. | |
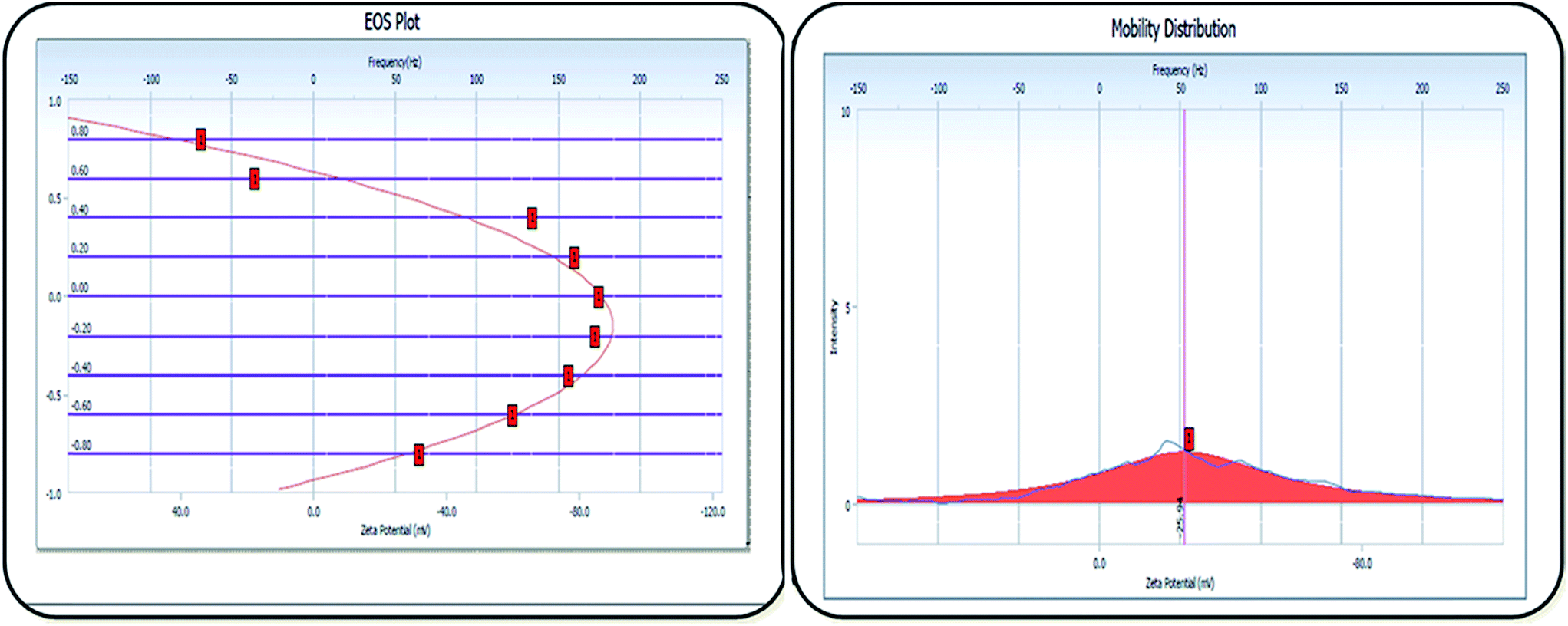
The zeta-potential value of ACE–β-CD-liposomes was found to be −25.94 mV (Fig. 6). The high negative value of the zeta-potential of the developed liposomal β-CD carriers signified high stability of the colloidal dispersion, which is a major contributor to the stability of such carriers.24,45
 |
| | Fig. 6 Zeta potential of ACE–β-CD-liposomes. | |
5.2.2. Confocal laser scanning microscopy (CLSM). CLSM studies depicted the spherical nature of the developed carrier systems (Fig. 7a). The studies were performed to evaluate the actual carrier functions of liposomes, by visualizing the distribution and penetration depth of the coumarin-6 fluorescent dye-entrapped β-CD liposomes into rat skin after 6 h of topical application exhibiting the transport potential of the carriers to the deeper layers of skin.25,46 Fig. 7b clearly reveals the passage of intact β-CD liposomes through intracellular channels of the skin. The study ratifies that the vesicular system can penetrate the intact skin layers,46 leading to permeation enhancement and delivery of the drug to the deep-sited targets in the skin.
 |
| | Fig. 7 (A) Confocal laser scanning photomicrograph of ACE–β-CD-liposomes (B) spherical fluorescent vesicular structures in skin layers. | |
5.2.3. Scanning electron microscopy (SEM). The three-dimensional nature of ACE–β-CD-liposomes was evident by SEM as depicted in Fig. 8. Scanning electron micrographs confirmed that ACE–β-CD liposomes possessed a spherical shape.29
 |
| | Fig. 8 SEM photomicrographs of ACE–β-CD-liposomes and inset shows magnified view of single vesicle. | |
5.2.4. Vesicle count. The density of vesicle population of un-sonicated ACE–β-CD liposomes was found to be 5.0 (±0.19) × 104 mm3. This confirmed the formation of vesicles in sufficient number to entrap the drug molecules.47
5.2.5. pH measurement. The pH of the final formulation was found to be 6.6 which is close to the pH of normal skin (5.6–6.4). Hence, the developed system plausibly is non-irritant in nature.
5.2.6. Fourier transform infra-red spectroscopy (FT-IR). The spectra of ACE, β-CD complex, PL 90G and ACE–β-CD-liposomal formulation is portrayed in Fig. 9. As depicted in Fig. 9, the IR spectrum of the developed formulation is entirely different from drug and PL. Major peaks of the ACE were either masked or shifted in β-CD-liposomes indicating significant intermolecular interactions. The principle peaks of ACE alone were observed at 3318 cm−1, 1770 cm−1, 1718 cm−1, 1446 cm−1, 1252 cm−1 and 1053 cm−1 that confirmed the purity of drug sample. Major peaks of β-CD complex were observed at 3378 cm−1, 2925 cm−1 and 1649 cm−1 and for PL were observed at 3010 cm−1, 2925 cm−1 and 1737 cm−1. The IR spectrum of drug with excipients shows the characteristic peaks with insignificant shift and reduced intensity. This ratifies the absence of any interaction between drug and excipients.42,43
 |
| | Fig. 9 FT-IR spectra of ACE, β-CD complex, PL 90G, ACE–β-CD-liposomal formulation. | |
5.2.7. Rheological studies. The developed ACE–β-CD liposomal gel exhibited non-Newtonian behaviour with significant magnitude of the yield value. Table 1 enlists the various rheological attributes of the developed gels, as per the Herschel–Bulkley model. The value of ‘n’ was found to be less than 1, for both the studied systems i.e., β-CD liposomal gel and MKT gel formulation, hence the system was found to be shear thinning in nature. The viscosity of the prepared liposomal gel was found to be higher than the MKT gel, thereby, indicating greater contact time of the former with the skin, hence higher therapeutic effect.15,24
Table 1 Values of power-law exponent (n), consistency index (K) and yield value of the developed ACE–β-CD-liposomal and MKT gel
| Formulation |
n |
K (Pa sn) |
Yield value (Pa) |
| ACE–β-CD-liposomal gel |
0.719 ± 0.019 |
4.66 |
3.625 |
| MKT gel |
0.2962 ± 0.101 |
122.75 |
1.278 |
5.2.8. Texture analysis (spreadability). Fig. 10a and Table 2, reveal that the developed ACE–β-CD liposomal gel exhibited fairly good gel strength, ease of spreading and extrusion from tubes as compared to MKT gel (Fig. 10b). The developed ACE–β-CD liposomal gel possessed an adequate cohesiveness, which is essential to hold the formulation at the site of application appreciably and will release its maximum drug to the dermal layers. Further, uniformity of curve, plotted using the Exponent 32® software, confirmed the smoothness and absence of any grittiness or lumps in the β-CD liposomal hydrogel. Developed gels, thus, met the criteria of absence of particulate matter and grittiness, as desired for any topical preparation.31
 |
| | Fig. 10 (A) Textural analysis of the formulated ACE–β-CD-liposomal gel (B) MKT gel. | |
Table 2 Various parameters obtained from the texture analysis of ACE–β-CD liposomal and MKT gel
| Formulations |
Firmness (g) |
Work of shear (g sec) |
Stickiness (g) |
Work of adhesion (g sec) |
| ACE–β-CD-liposomal gel |
148 |
120.8 |
92 |
84.2 |
| MKT gel |
272.4 |
220.64 |
185.6 |
122.4 |
5.3. Ex vivo drug permeation studies
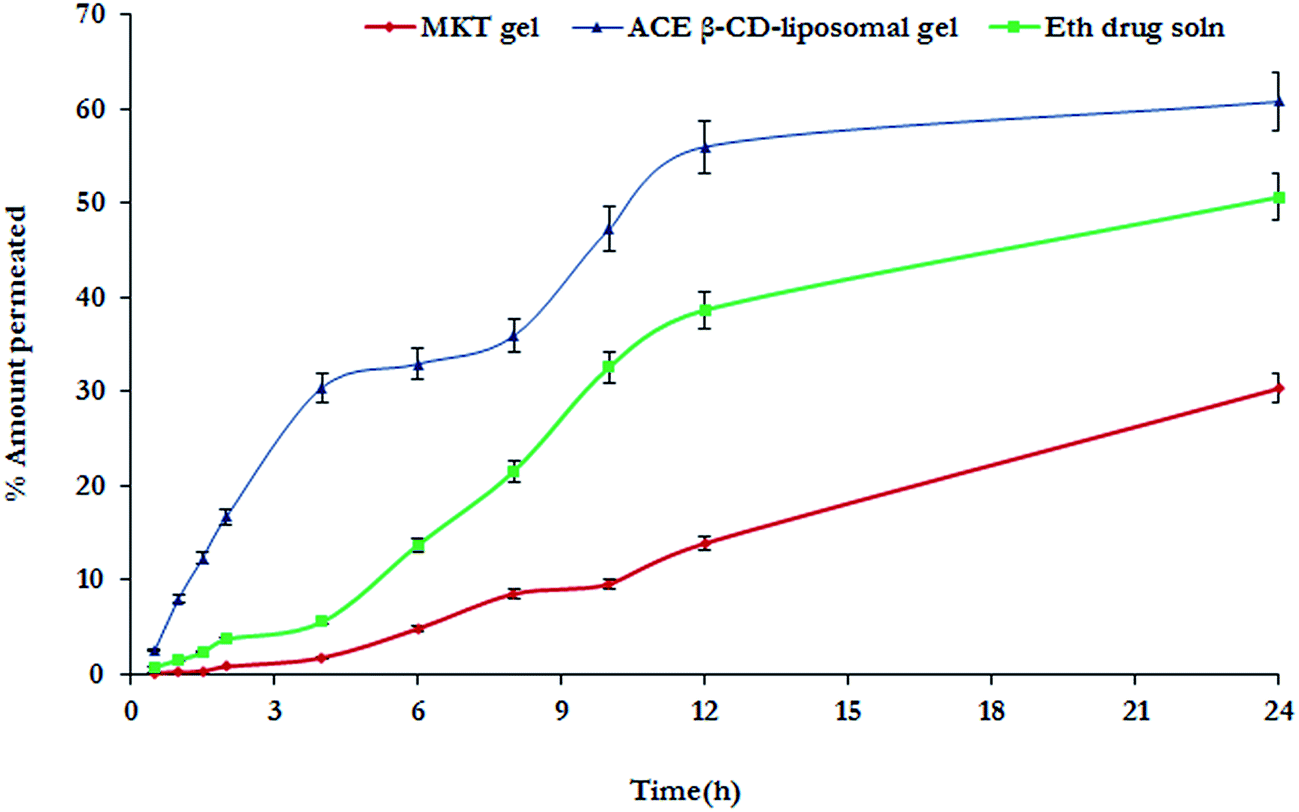
The permeation profile of ACE from various developed formulations employing LACA mice skin is depicted in Fig. 11. It is vivid from the results that the drug transport characteristics of both of optimized ACE–β-CD liposomal gel (61%) was found to be better than the MKT gel (30%) and ethanolic drug solution (51%). This observation is in accord to the reported superiority of the novel carrier-based formulations vis-à-vis the MKT gel formulation. The better drug transport characteristics of the vesicular carriers can be attributed to the interaction of the biocompatible components i.e., phospholipids with the skin resulting in better penetration. The results of skin retention on the other hand, reveal that the MKT gel formulation exhibits 1.2 times greater retention than that of ethanolic drug solution and 2.8 times greater than ACE–β-CD liposomal gel. This may be accounted to the superiority of the vesicular carriers due to their high permeation properties within the layers of dermis by virtue of the integration of PLs to the skin lipids (similar in nature). The order of skin retention was observed to be: MKT gel (15%) > ethanolic drug soln (13%) > ACE–β-CD liposomal gel (8%) as depicted in Fig. 12. Despite the presence of phospholipids and the vesicular nature of liposome, these systems offered lower values of skin retention due to presence of alcohol, which might have resulted in dermal extraction of drug too.24,27
 |
| | Fig. 11 Plot between mean percentage of ACE permeated versus time from various formulations. Each cross bar indicates average value ± SD (n = 3). | |
 |
| | Fig. 12 Bar diagram showing the mean percent drug retained in skin from various formulations. Each cross bar indicates average value ± SD (n = 3). | |
5.4. In vitro drug deposition studies in dermal layers: dermatokinetic modelling
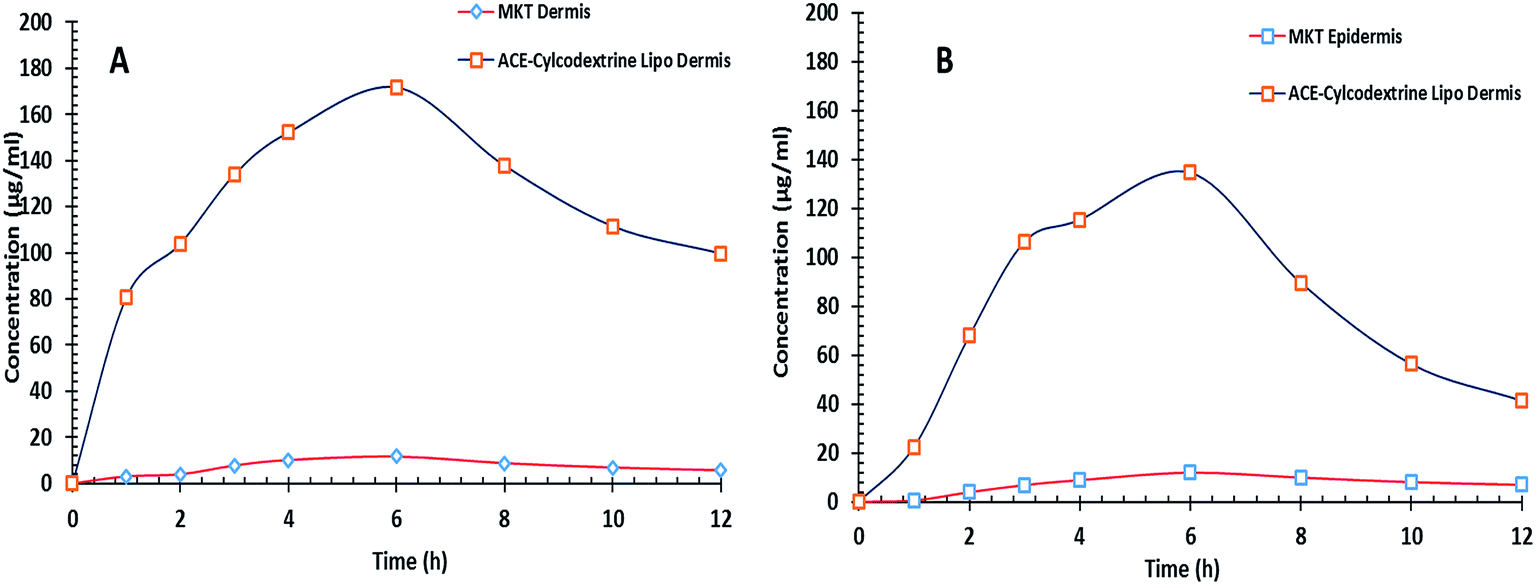
Fig. 13a and b show the variation of ACE concentration in dermis and epidermis of the LACA mice after single application of various ACE formulations. The delivery of ACE in the skin layers by ACE–β-CD liposomal gel was found to be significantly greater (p < 0.05) than the MKT gel. Table 3 gives the numeric values of AUC0–12h, Cskinmax, skin penetration rate constant (Kp), Tskinmax and skin elimination rate constant (Ke). It is vivid from the results that the biological half-life of the drug was enhanced by ACE–β-CD-liposomes in both the skin layers and Tmax was reduced drastically. Cmax, in both the layers and AUC in dermis was significantly enhanced. The findings indicate the existence of non-concordance between the results obtained from skin retention protocol. This can be ascribed to the difference in the design and model of both the experiments. The drug retention in skin was a single point study till 6 h, whereas the deramtokinetic protocol was a multiple point study, which is extended till 12 h. From the dermatokinetic data, it can be seen that the Tmax value for the MKT product was very close to the sampling time, i.e., 6 h, whereas for the developed system, it was almost 1 h post Tmax. In the present situation, the momentarily display of drug deposited in skin in the form of drug retention can be seen as a biased end point towards MKT product. On the other hand, the dermatokinetic profile provided compete time-dependent unbiased sojourn of drug in two layers of skin. Henceforth, it can be concluded that ACE–β-CD liposomes possesses the potential to enhance the delivery of ACE and increase its topical bioavailability vis-à-vis the MKT product.25,32
 |
| | Fig. 13 Dermatokinetic profile showing the drug concentration in: (A) dermis; and (B) epidermis, at various time points. Each cross bar indicates average value ± SD (n = 3). | |
Table 3 Dermatokinetic parameters (mean ± SD) of ACE loaded topical formulations in epidermis and dermis (n = 3)
| Parameters |
MKT gel (mean ± SD) |
ACE–β-CD-liposomal gel (mean ± SD) |
| Epidermis |
Dermis |
Epidermis |
Dermis |
| AUC0–12h (μg cm−1 h−1) |
208.99 ± 7.02 |
149.89 ± 6.19 |
1332.28 ± 32.84 |
2318.38 ± 29.14 |
| Cskinmax (μg cm−1) |
8.93 ± 1.29 |
8.72 ± 1.01 |
103.18 ± 5.27 |
158.67 ± 9.39 |
| Tskinmax (h) |
6.3 ± 0.421 |
5.71± 0.12 |
5.01 ± 0.29 |
5.16 ± 0.05 |
| Ke (h−1) |
7.20 ± 0.05 |
4.0 ± 0.10 |
3.03± 0.67 |
2.40 ± 0.17 |
| Kp (h−1) |
4.01± 0.03 |
3.99 ± 0.08 |
3.57 ± 0.07 |
4.54 ± 0.12 |
5.5. Stability studies
As discerned from the tabulated results (Table 4), the developed ACE–β-CD liposomal gel formulation was found to be stable at all the studied storage conditions. However, slight decrease in the assay as well as alteration in the formulation attributes was observed at high temperature after 90 days. Storage at refrigerated conditions showed remarkably better stability of the formulation for the studied period. After 90 days at high temperature (40 ± 2 °C/75 ± 5% RH), the drug leakage from ACE–β-CD liposomal gel was up to the tune of 11.72% compared to 2.11% at refrigerated conditions (5 ± 3 °C). The results revealed 5.5 times higher drug leakage (i.e., 11.72%) from ACE–β-CD liposomal gel at higher temperature (Fig. 14a and b) vis-à-vis the formulation stored at 5 ± 3 °C i.e., (2.11%). The leakage of the drug from the vesicles stored at higher temperature may be assigned to the effect of the temperature on the gel to liquid transition of lipid bilayers together, eventually altering their packing.15,48,49 These results indicate that the optimized ACE–β-CD-liposomal system should be preferably stored at room temperature or at refrigerated temperature.48
Table 4 Stability testing of the developed formulation of ACE–β-CD liposomal gel stored at various temperatures (n = 3)
| Days |
5 ± 3 °C |
40 ± 2 °C/75 ± 5% RH |
| 0 |
101.73 ± 0.86 |
101.73 ± 0.54 |
| 15 |
100.13 ±0.56 |
99.92 ± 0.18 |
| 30 |
99.89 ± 0.68 |
97.39 ± 0.26 |
| 60 |
99.71 ±0.56 |
94.12 ± 0.95 |
| 90 |
99.62 ± 0.89 |
95.01 ±0.16 |
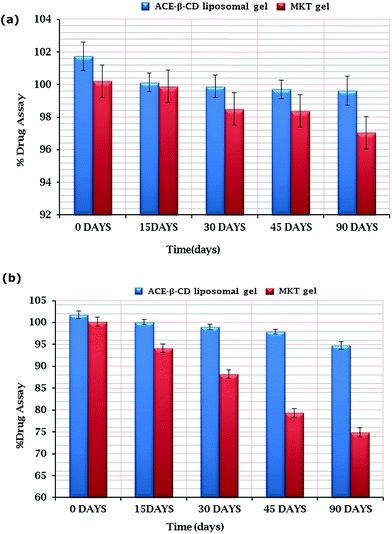 |
| | Fig. 14 (a) Bar diagram depicting the % drug assay at different time intervals for ACE–β-CD-liposomal gel and marketed gel at 5 ± 3 °C. (b) Bar diagram depicting the % drug assay at different time intervals for ACE–β-CD-liposomal gel and marketed gel at 40 ± 2 °C/75 ± 5% RH. | |
5.6. Skin compliance studies
As revealed by the histopathological studies, ACE–β-CD liposomal gel showed almost no skin erythema, whereas high redness scores were observed in case of MKT gel as depicted in Table 5 and in the actual photographs (Fig. 15). In case of MKT gel, the irritation scores increased day-by-day whereas the application of liposomal gel showed almost no signs of irritation.35
Table 5 Mean erythemal score observed for various groups: (A) untreated, (B) MKT gel formulation and (C) ACE–β-CD-liposomal gel (n = 3)
| Group |
Mean erythemal score |
| Days |
| 1 |
2 |
3 |
4 |
5 |
6 |
7 |
| A |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
| B |
1 |
1 |
1 |
1 |
1 |
2 |
2 |
| C |
0 |
0 |
0 |
0 |
0 |
0 |
1 |
 |
| | Fig. 15 Skin safety studies at the end of seven days along with their histopathological photomicrographs: (A) untreated; (B) treated with conventional gel and (C) treated with ACE–β-CD-liposomal gel (magnification 100×). | |
Further, histopathological evaluation revealed that the developed system was well-tolerated on mouse skin vis-à-vis the MKT product, as no signs of disruption and irritation were observed visually as well as microscopically. The groups treated with ACE–β-CD-liposomal gel did not show any disruption in the integrity of the normal skin and also did not show any signs of inflammation while marked disruption and inflammation was observed in the photomicrographs of the mice skin treated with the MKT product (Fig. 15). Hence, the current study supports the hypothesis of biocompatible nature of the developed formulation and hence, the formulation was well tolerated by mouse skin.27,31
5.7. Anti-inflammatory activity
5.7.1. Carrageenan-induced paw oedema in rats. The degree of inflammation (i.e., paw swelling) and inhibition of oedema (i.e., drug activity) is demonstrated in Fig. 16a and b for the carrageenan-induced rat paw oedema. In the control group, a rapid and continuous increase in paw volume (i.e., oedema) was observed and the inflammation was sustained during the entire period of 24 h of the study. In the groups receiving active formulations (i.e., ACE–β-CD liposomal gel, MKT gel formulation), the percentage increase in paw volume was lower vis-à-vis control groups (plain gel), indicating that all tested formulation of ACE possess anti-inflammatory activity. The inflammation due to carrageenan was markedly inhibited by the ACE–β-CD liposomal gel. The percentage inhibition was found to be much higher at every time point with ACE–β-CD liposomal gel (p < 0.001). The results of the study revealed the advantage of the vesicular carriers over the conventional systems. This can be ascribed to their better interaction with the skin components and skin-depot forming potential.37–39
 |
| | Fig. 16 (A) Shows the curves for the % paw swelling in MKT gel and ACE–β-CD-liposomal gel after carrageenan treatment. (B) Shows the curves for the % edema inhibition in MKT gel and ACE–β-CD-liposomal gel after carrageenan treatment. | |
5.7.2. Tail-flick method. The results in Fig. 17 are indicative of the comparative studies between the analgesia produced by ACE–β-CD liposomal gel and the MKT gel. Percent analgesia caused by ACE-loaded liposomal formulation was significantly greater (p < 0.001) than of the MKT gel.
 |
| | Fig. 17 Shows the bar diagram depicting % analgesic effect produced by ACE–β-CD-liposomal gel in comparison with the MKT gel formulation. | |
MKT gel formulation offered shorter onset of action, while duration of action was found to be prolonged for the liposomal formulation. This was due to fact that ACE was present within the vesicular systems, due to which duration of action was prolonged, but in case of MKT formulation, drug was not encapsulated but present in the free from due to which it offered shorter duration of action. However, the prolonged duration of action was a result of better dermal retentivity of drug by liposomes. Also, the observed enhanced analgesic effect by use of biocompatible PLs is in agreement with the previous reports.29,31
6. Conclusion
In conclusion, the work as envisaged with the aim of improvement in the delivery as well as stability by exploiting the potential of novel carrier systems, could achieve its targeted benefits. The twin challenges of the drug ACE, relating to inappropriate physicochemical properties (as responsible for insolubility) and hydrolytic cleavage (responsible for the loss of drug in aqueous environment of the novel systems like liposomes) have really been a tough task to accomplish. Thus, this strategy of combining the two different approaches which otherwise interestingly, share the same philosophy of supramolecular association through non-covalent bondings, and have paved the way for a formulation of ACE for topical application with optimum efficacy, safety, stability and compliance. The outcome of these stability studies, therefore, further proposes for the extension of the work to be taken for mass level of utilisation by supplementing and complimenting with investigations as desired.
Conflict of interest
Authors report no conflict of interest.
Acknowledgements
Authors are thankful to University Grants Commission (UGC), New Delhi, India, M/s Ipca Labs Ltd., Mumbai, India for research grant and M/s Phospholipid GmbH, Nattermannallee, Germany, for the ex-gratis supply of phospholipids.
References
- R. N. Brogden and L. R. Wiseman, Drugs, 1996, 52, 113–124 CrossRef CAS PubMed.
- N. G. R. Rao, U. Kulkarni, A. Deshmukh and D. K. Suresh, Int. J. Pharm. Sci. Drug Res., 2010, 2, 107–111 CAS.
- K. M. Manjanna, B. Shivakumar and T. M. Pramod, Crit. Rev. Ther. Drug Carrier Syst., 2010, 27, 509–545 CrossRef CAS PubMed.
- R. H. Jones and C. L. Tait, Br. J. Clin. Pract., 1995, 49, 67–70 CrossRef CAS PubMed.
- U. A. Boelsterli, Drug Saf., 2002, 25, 633–648 CrossRef CAS PubMed.
- G. M. Shingade, J. Drug Deliv. Ther., 2012, 2, 66–76 CAS.
- O. P. Katare, K. Raza, B. Singh and S. Dogra, Indian J. Dermatol. Venereol. Leprol., 2010, 76, 612–621 CrossRef PubMed.
- K. Raza, O. P. Katare, A. Setia, A. Bhatia and B. Singh, J. Microencapsulation, 2013, 30, 225–236 CrossRef CAS PubMed.
- K. Raza, B. Singh, P. Singal, S. Wadhwa and O. P. Katare, Colloids Surf., B, 2013, 105, 67–74 CrossRef CAS PubMed.
- E. Legrand, Expert Opin. Pharmacother., 2004, 5, 1347–1357 CrossRef CAS PubMed.
- R. Yamazaki, S. Tawai, T. Matsuzaki, N. Kaneda, S. Hashimoto, T. Yokokura, R. Okamoto, T. Koshino and Y. Mizushima, Eur. J. Pharmacol., 1997, 329, 181–187 CrossRef CAS PubMed.
- C. S. Kumar, P. Yesupadam, L. Lakshmi and C. Sekhar, In. J. Pharm., 2010, 1, 23–27 Search PubMed.
- M. Dooley, C. M. Spencer and C. J. Dunn, Drugs, 2001, 61, 1351–1378 CrossRef CAS PubMed.
- K. Raza, M. Kumar, P. Kumar, R. Malik, G. Sharma, M. Kaur and O. P. Katare, BioMed Res. Int., 2014, 2014, 406731 Search PubMed.
- A. Bhatia, K. Raza, B. Singh and O. P. Katare, J. Cosmet., Dermatol. Sci. Appl., 2009, 8, 282–284 CrossRef PubMed.
- J. R. Bhinge, R. V. Kumar and V. R. Sinha, J. Chromatogr. Sci., 2008, 46, 440–444 CAS.
- F. H. Silver, J. W. Freeman and D. DeVore, Skin Res. Technol., 2001, 7, 18–23 CAS.
- E. M. Martin Del Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef.
- K. L. Yap, X. Liu, J. C. Thenmozhiyal and P. C. Ho, Eur. J. Pharm. Sci., 2005, 25, 49–56 CrossRef CAS PubMed.
- T. Higuchi and A. Connors, Phase Solubility Techniques, 1965, 4, 117–211 CAS.
- G. Blume and G. Cevc, Biochim. Biophys. Acta, 1990, 1029, 91–97 CrossRef CAS.
- N. Kaur, R. Puri and S. K. Jain, AAPS PharmSciTech, 2010, 11, 528–537 CrossRef CAS PubMed.
- S. K. Jain, Y. Gupta, A. Jain and M. Bhola, Drug Delivery, 2007, 14, 327–335 CrossRef CAS PubMed.
- P. Negi, B. Singh, G. Sharma, S. Beg and O. P. Katare, J. Microencapsulation, 2015, 32, 419–431 CrossRef CAS PubMed.
- P. Negi, B. Singh, G. Sharma and O. P. Katare, J. Nanopharmaceutics Drug Delivery, 2014, 2, 1–10 CrossRef PubMed.
- J. Shim, H. Seok Kang, W. S. Park, S. H. Han, J. Kim and I. S. Chang, J. Controlled Release, 2004, 97, 477–484 CrossRef CAS PubMed.
- K. Raza, B. Singh, S. Lohan, G. Sharma, P. Negi, Y. Yachha and O. P. Katare, Int. J. Pharm., 2013, 456, 65–72 CrossRef CAS PubMed.
- S. Jain, P. Jain, R. B. Umamaheshwari and N. K. Jain, Drug Dev. Ind. Pharm., 2003, 29, 1013–1026 CrossRef CAS PubMed.
- K. Raza, M. A. Shareef, P. Singal, G. Sharma, P. Negi and O. P. Katare, J. Liposome Res., 2014, 24, 290–296 CrossRef CAS PubMed.
- F. Maestrelli, M. L. Gonzalez-Rodriguez, A. M. Rabasco and P. Mura, Int. J. Pharm., 2005, 298, 55–67 CrossRef CAS PubMed.
- P. Negi, B. Singh, G. Sharma, S. Beg, K. Raza and O. P. Katare, Drug Delivery, 2015, 1–17 Search PubMed.
- K. Raza, B. Singh, S. Singla, S. Wadhwa, B. Garg, S. Chhibber and O. P. Katare, Mol. Pharm., 2013, 10, 1958–1963 CrossRef CAS PubMed.
- A. M. Kligman and E. Christophers, Arch. Dermatol., 1963, 88, 702–705 CrossRef CAS.
- ICH, 2003.
- J. Draize, G. Woodard and H. Calvery, J. Pharmacol. Exp. Ther., 1944, 82, 377–390 CAS.
- S. K. Kulkarni, Handbook of Experimental Pharmacology, Vallabh Prakashan, Delhi, 3rd edn, 1999 Search PubMed.
- C. A. Winter, E. A. Risley and G. W. Nuss, Proc. Soc. Exp. Biol. Med., 1962, 111, 544–547 CrossRef CAS PubMed.
- P. R. Patil, T. Bera and V. M. Patil, J. Pharm. Biomed. Sci., 2010, 4, 1–5 Search PubMed.
- R. Vinegar, J. F. Traux and J. L. Selph, Fed. Proc., 1987, 46, 118–126 CAS.
- M. M. Ibrahim, M. El-Nabarawi and M. A. Fadlalla, AAPS PharmSciTech, 2010, 11, 1730–1737 CrossRef CAS PubMed.
- A. A. Elzainy, X. Gu and F. Simons, AAPS J., 2004, 6, 18 CrossRef PubMed.
- D. R. Patil, P. G. Ingole, K. Singh and D. S. Dalal, Res. J. Chem. Sci., 2012, 2, 60–63 CAS.
- J. Rupal, J. Kaushal, S. C. Mallikarjuna and P. Dipti, Int. J. Pharm. Sci., 2009, 1, 32–35 CAS.
- S. J. George and D. T. Vasudevan, Pharmaceutics, 2012, 4, 220–227 CAS.
- K. Kostarelos, P. F. Luckham and T. F. Tadros, J. Chem. Soc., Faraday Trans., 1998, 94, 91–145 RSC.
- K. Ahmed, P. N. Gribbon and M. N. Jones, J. Liposome Res., 2002, 12, 285–300 CrossRef CAS PubMed.
- K. Raza, B. Singh, A. Mahajan, P. Negi, A. Bhatia and O. P. Katare, J. Drug Targeting, 2011, 19, 293–302 CrossRef CAS PubMed.
- R. M. Lopez, A. Ayestaran, L. Pou, J. B. Montoro, M. Hernandez and I. Caragol, Am. J. Health-Syst. Pharm., 1996, 53, 2724–2727 CAS.
- K. Vorauer-Uhl, K. Wagner and H. Katinger, Eur. J. Pharmacol., 2002, 43, 47 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Kanika Thakura and
O. P. Katare*a
b,
Kanika Thakura and
O. P. Katare*a