
Starch nanoparticles prepared in a two ionic liquid based microemulsion system and their drug loading and release properties
Abstract
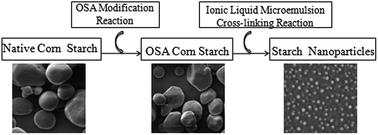
In this work, 1-hexadecyl-3-methylimidazolium bromide ([C16mim]Br) and 1-octyl-3-methylimidazolium acetate ([C8mim]Ac) were simultaneously used as substitutes for surfactants and the polar phase to prepare [C16mim]Br/butan-1-ol/cyclohexane/[C8mim]Ac ionic liquid microemulsions. Then, the structure of the microemulsion was investigated by pseudo-ternary phase diagram, dynamic light scanning (DLS) and conductivity measurement. Starch nanoparticles with a mean diameter of 80.5 nm were prepared with Octenyl Succinic Anhydride (OSA) starch as raw material through ionic liquid-in-oil (IL/O) microemulsion cross-linking reaction. Scanning electron microscope (SEM) data revealed that starch nanoparticles were spherical granules with small size. In addition, the particles presented homogeneous distribution and no aggregation phenomenon appeared. The results of Fourier transform infrared spectroscopy (FTIR) identified the formation of cross-linking bonds in starch molecules. Finally, the drug loading and releasing properties of starch nanoparticles were investigated with mitoxantrone hydrochloride as a drug model. This work might provide an efficient method to synthesis starch nanoparticles.
 Please wait while we load your content...
Please wait while we load your content...

