
A theoretical study of dirhodium-catalyzed intramolecular aliphatic C–H bond amination of aryl azides†
Abstract
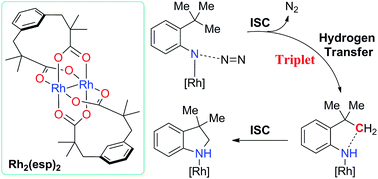
Dirhodium-contained catalysts mediated aliphatic C–H bond amination of aryl azides were studied using BPW91 functional. Calculations show the reactions with Rh2(esp)2 (esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid) and the model compound Rh2(OCHO)4 as catalysts take place via similar mechanisms. Firstly, the dirhodium metal complex coordinates with the substrate and releases nitrogen gas. This rate-determining step results in the formation of the metal nitrene. Metal nitrene mediated intramolecular C–H bond amination is conducted via two alternative pathways, respectively. The singlet metal nitrene mediated intramolecular C–H bond amination occurs via a concerted and asynchronous pathway involving direct metal nitrene insertion into the C–H bond. The triplet metal nitrene case is a stepwise pathway involving a hydrogen transfer and then a diradical recombination. Our study suggests the triplet H-abstraction is more favorable than the singlet one. The resulted triplet intermediate would not go through the high-barrier diradical recombination process, but across to the singlet pathway via a MECP and form the final singlet product. The tethered esp ligands in Rh2(esp)2 provide steric effects to constrain the substrate-catalyst compound but indicates inconspicuous influence on the mechanisms of dirhodium catalyzed aliphatic C–H bond amination of aryl azides.
 Please wait while we load your content...
Please wait while we load your content...

