
PVP-derived carbon nanofibers harvesting enhanced anode performance for lithium ion batteries
Abstract
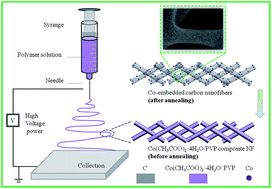
Co-embedded carbon nanofibers were synthesized using electrospinning with polyvinylpyrrolidone (PVP) instead of high cost polyacrylonitrile (PAN). The obtained composite nanofibers as an anode material for lithium ion batteries deliver a reversible capacity of 542.6 mA h g−1 in the 100th cycle at a current density of 100 mA g−1. Moreover, the anode material shows better cycle performance and rate capability in comparison to the resultant product without Co additive. It is believed that the significant improvement is attributed to the nanofiber morphology with high surface-to-volume ratio as well as the existence of Co nanoparticles that enhance the electrical conductivity of the nanocomposites.
 Please wait while we load your content...
Please wait while we load your content...

