DOI:
10.1039/C5RA23830G
(Paper)
RSC Adv., 2016,
6, 15513-15517
Polyoxometalate-based supramolecular architecture constructed from a purely inorganic 1D chain and a metal–organic layer with efficient catalytic activity†
Received
11th November 2015
, Accepted 28th January 2016
First published on 1st February 2016
Abstract
A new polyrotaxane structure, {Ag8O(Htrz)4(4,4′-bpy)2}{AgPMo12O40} (Htrz = 1,2,4-triazole, 4,4′-bpy = 4,4′-bipyridine) (1), was obtained by the reaction of Ag+, H5PMo10V2O40 and mixed organic linking ligands under hydrothermal conditions. The structure has been characterized by IR, TG analysis, elemental analysis, X-ray powder diffraction and single-crystal X-ray diffraction. In 1, a purely inorganic 1D chain composed of Keggin-type polyoxoanions and Ag+ linkers, interluded into the 2D Ag–organic layers, resulting in the polyrotaxane structure. This is the first polyrotaxane compound constructed from a POM-based purely inorganic 1D chain and Ag-Htrz-4,4′-bpy 2D networks. The ratio of the mixed ligands played a crucial role in the synthesis of the title compound. Moreover, compound 1 exhibited efficient catalytic activity for the oxidation of desulfurization, and possessed electrocatalytic activity for the reduction of H2O2 and BrO3−.
Introduction
Organic–inorganic hybrid materials have received more and more attention due to their structural diversity and a wide range of applications including catalysis, gas adsorption, pharmaceuticals, etc.1 Polyoxometalates (POMs), as a class of unique inorganic metal–oxygen clusters with a nano-size structure, have been explored as efficient functional components in many fields.2 In the past few decades, POMs were introduced into the inorganic–organic hybrid materials to importantly improve their property and expand the range of their applications, resulting in more and more innovative functional materials.3 In particular, the exploration of the POM-based metal–organic frameworks (PMOFs) has made them more charming.4 In the PMOFs, the POMs usually acted as the guest molecules to functionalize the metal–organic units. In this filed, the HUST, MIL-101, UiO and some other types of MOFs were frequently used as the molecular platform to encapsulate the POM guest molecules.5,6 In another aspect, POMs were used as the node to coordinate with the organic ligands and metal–organic groups for constructing the PMOFs.7 The emerging of these PMOFs has attracted more and more attention in introduction of the POMs into the 3D inorganic–organic hybrid materials.
Rotaxane molecule is a combination of one or more cyclic segments and the chain-like units, where the 1D chain passes through the cavity of cyclic molecule. If the structure contains multiple cyclic molecules, it will form a polyrotaxane framework, which represents a significant subclass of the organic–inorganic hybrid materials.8 As an excellent inorganic building blocks, many chemists have focused on introducing the POMs into the polyrotaxane structure. For instance, Wang et al. prepared a novel polyrotaxane framework, [CuII(L)2(H2O)2][CuI2(L)2]PMo12O40 (L = 4,4-bis(1,2,4-triazol-1-ylmethyl)biphenyl) in 2007, in which the isolated Keggin anion was encapsulated into the framework as the guest molecule.9a In 2009, Su et al. used the [β-Mo8O26]4− as the guest forming an attractive 2D polythreading topology [CuI3(L)2(Mo8O26)0.5Cl] (L = 3-((1H-1,2,4-triazol-1-yl)methyl)pyridine).9b Noteworthy, Wang and co-workers obtained two fascinating isostructural polyrotaxane frameworks, [Cu2(L)3(SiMo12O40)·(H2O)6] and [Cu2(L)3(SiW12O40)·(H2O)6] (L = N,N′-bis(3-pyridinecarboxamide)-1,4-butane), where the Keggin-type polyoxoanions acted as the templates sandwiched by two adjacent 2D polyrotaxane sheets.9c In this paper, we firstly reported a polyrotaxane structure based on the metal–organic 2D sheets and purely inorganic 1D POM chain {Ag8O(Htrz)4(4,4′-bpy)2}{AgPMo12O40} (1). The 1D POM-based chain was composed of the Keggin-type POMs and Ag+ linkers, and the 2D sheets consisted of Ag+ cations and two N-donor ligands. Electrocatalytic study indicated that it possessed of electrocatalytic activity for reduction of H2O2 and BrO3−, and the title compound exhibited efficient catalytic activity for the desulfurization.
Results and discussion
Synthesis and structure
Compound 1 was prepared from the mixture of H5PMo10V2O40 ({PMo10V2}), Htrz, 4,4-bpy and AgNO3 under the hydrothermal conditions. During the reaction, the use of {PMo10V2} is a critical step for the synthesis of 1, and the conversion of {PMo10V2} to {PMo12} has happened in the reaction. In the synthesis, we have tried to use the {PMo12} as the start materials to replace the {PMo10V2}, the resulting product (4,4-bpy)]4[H3PMo12O40]·5H2O was composed of four free 4,4-bpy ligands around a {PMo12} anion (Fig. S1, Table S1†). Further, the use of the mixed ligands Htrz and 4,4′-bpy was also play an important role in the isolation of the polyrotaxane structure. The ratio of Htrz and 4,4′-bpy should be in the range of 2.8–28 for obtaining the title compound, and was optimized as 2.8–3.0 to get the maximum yield of pure crystals.
Compound 1 crystallized in the tetragonal space group I4/mcm, and possessed of a polyrotaxane structure consisting of the porous 2D metal–organic layers (Fig. 1a) and purely inorganic 1D chain (Fig. 1b). As shown in Fig. 1, the purely inorganic 1D chain interludes in the porous 2D metal–organic layers forming the 3D polyrotaxane structure. In the 2D porous layer, there are two linking units, one square Ag4O(Htrz)4 (Fig. 2a) unit, which was composed of four Ag+ ion, four Htrz ligands and a central oxygen atom. In this unit, each Ag+ cation coordinated with two nitrogen atoms from two Htrz ligands and a central oxygen atom, and each Htrz ligand coordinated with two Ag+ ions, forming the square building units. The other one is a line-like linker, composed of two Ag+ ions and a 4,4′-bpy ligand. In the 2D layer, each square Ag4O(Htrz)4 was connected with the neighboring four Ag4O(Htrz)4 units via four Ag2(4,4-bpy) linkers forming the 4-connected 2D porous layer with the pore size of 21.49 × 21.49 Å2. The Ag–N bond lengths are in the range of 1.912(17) Å to 2.39(3) Å. The pore size is large enough to encapsulate one Keggin polyoxoanion. As shown in Fig. 1b, the Keggin-type anions {PMo12} and Ag+ cations are alternately connected to form purely inorganic 1D chain, where each Ag+ ion was coordinated by eight oxygen atoms. The purely inorganic 1D anionic chain and the 2D metal–organic cationic layers are stacked together via the static interactions, resulting the electroneutral 3D supermolecular materials.
 |
| | Fig. 1 (a) Ball-and-stick representation of the 2D layer in the polyrotaxane structure; (b) ball-and-stick representation of purely inorganic 1D chain; (c) ball-and-stick representation of 3D polyrotaxane structure. | |
 |
| | Fig. 2 Ball-and-stick representation of the (a) square building unit and the (b) liner unit in the 2D layer; (c) ball-and-stick representation of the 2D layer in the polyrotaxane structure. | |
Oxidative desulfurization
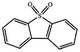
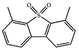
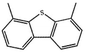
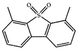
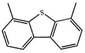
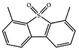
Currently, the environmental pollution has received more and more attention, especially for the sulfur dioxide pollution. So a significant task is the desulfurization of fossil fuels, which is closely related to people's living environment.10 In this research area, the exploration of the efficient catalysts for oxidative desulfurization has become a recognized effective strategy for the desulfurization of fossil fuels. Here we performed the oxidation of thiophene to sulfone as the test reaction to identify the catalytic activity of this title compound. The typical process of the oxidation of dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) in the presence of compound 1 were carried out in CH2Cl2 at 60 °C by using tert-butyl hydroperoxide (TBHP) as the oxidant (Scheme S1†). The maximum conversion of substrates were 99.8% after 120 min for DBT and 99.1% after 200 min for 4,6-DMDBT. For Na3PMo12 as the heterogeneous catalyst, the maximum conversion of the substrates were 88.1% after 120 min for DBT and 86.54% after 200 min for 4,6-DMDBT (Table 1). Both the substrates were ultimately oxidized into the corresponding sulfones with 100% selectivity. These results were confirmed by FTIR (Fig. S3†) and GC-MS (Fig. S4†) measurements. For the blank experiments, the conversion of substrates were 35.6% for DBT and 26.8% for 4,6-DMDBT. The experimental results indicated that 1 was an excellent catalyst for the oxidation of the sulfur-containing substrates (Fig. S5 and S6†). Further, compound 1 can be easily recovered from the reaction vessel by a centrifugation process. Powder X-ray diffraction (PXRD) studies revealed that the PXRD pattern of the recovered sample matching well with that of the calculated pattern from single-crystal diffraction data, and the IR spectrum of the recovered catalyst is almost identical to that of the as-synthesized sample (Fig. S7 and S8†). These results revealed that the catalyst is stable in this catalytic reaction. Additionally, we found that its catalytic activity could maintain in the recycled experiments, no significant loss in catalytic activity was observed (Fig. S9†). As shown in Table S2,† it could found that the title compound needed a shorter reaction time for achieving a similar percent conversion, although a relatively small amount of oxidant was used in this work.
Table 1 Oxidation of sulfide to sulfone with TBHP catalyzed by catalyst 1a
| Entry |
Substrate |
Product |
Catalyst |
t [min] |
Removalb [%] |
| Conditions: a mixture of catalyst (1.5 μmol), sulfide (80 μmol), and TBHP (240 μmol, 3 equiv.) in CH2Cl2 (2 mL) was stirred at 60 °C. Based on HPLC analysis. |
| 1 |
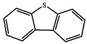 |
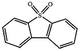 |
1 |
120 |
99.8 |
| 2 |
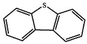 |
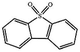 |
Na3PMo12 |
120 |
88.1 |
| 3 |
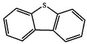 |
 |
None |
120 |
35.6 |
| 4 |
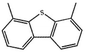 |
 |
1 |
200 |
99.1 |
| 5 |
 |
 |
Na3PMo12 |
200 |
86.54 |
| 6 |
 |
 |
None |
200 |
26.8 |
Electrochemical and electrocatalytic properties
POMs, as an important subclass of metal–oxygen nanoclusters, possess of empty orbitals in their structures. They can undergo a stepwise multi-electron reversible redox reaction without changing their original structure, and have exhibited interesting electrochemical and electrocatalytic properties.11 In this article, compound 1 was used to prepare the carbon paste electrode (CPE) for the study of its electrochemical property. As well known, the CPE is inexpensive and easy to be prepared and renewed.12 The cyclic voltammogram (CV) curve of 1-CPE was performed in 0.4 M HAc–NaAc buffer solution. As shown in Fig. 3a, in the potential range of −0.6 V to 0.6 V, three pairs of redox peaks (I–I′, II–II′ and III–III′) with E1/2 = 0.268, −0.053 and −0.302 V (E1/2 = (Epa + Epc)/2) appeared, which corresponded to the redox process of molybdenum centers in 1.13 Further, the CV curves of 1-CPE were performed in pH 4 HAc–NaAc buffer solution (Fig. 3b) with different scan rates. With the increase of the scan rates from 5 to 50 mV s−1, the linearity relationship between the peak currents and the square root of the scan rate (Fig. S10†) indicates that the electrode reaction is controlled by diffusion process. With the increase of the scan rates from 100–300 mV s−1 the relationship between the peak currents and scan rates was liner (Fig. S11†), indicating that the electrochemical process is surface controlled.14
 |
| | Fig. 3 (a) CV of 1-CPE in 0.4 M HAc–NaAc buffer solution (pH 4) at the scan rate of 100 mV s−1; (b) CV of 1-CPE in 0.4 M HAc–NaAc buffer solution (pH 4) at different scan rates (from inner to outer: 5, 10, 20, 50, 75, 100, 125, 150, 200, 250, 300 mV s−1); (c) electrocatalytic reduction of H2O2 by 1-CPE and bare electrode in pH 4 0.4 M HAc–NaAc buffer solution. Scan rate: 50 mV s−1; (d) electrocatalytic reduction of BrO3− by 1-CPE and bare electrode in pH 4 0.4 M HAc–NaAc buffer solution. Scan rate: 50 mV s−1. | |
Previous study indicated that POMs have been widely used as the electrocatalysts for removing the contaminants in water, such as iodates, bromates, nitrites and hydrogen peroxide.11,15 Hence, the electrocatalytic properties of 1-CPE were investigated towards the reduction of hydrogen peroxide and sodium bromate in the pH 4 HAc–NaAc buffer solution. With the addition of different concentrations of H2O2 and NaBrO3, the reduction peak current increased sharply while the corresponding oxidation peak current decreased. Also, no reduction of H2O2 and NaBrO3 could be observed in the absence of 1 in the explored potential domain. As a comparison, blank electrode does not show catalytic activity at a max concentration of H2O2 and NaBrO3. These results revealed that compound 1 displays electrocatalytic activity toward reduction of H2O2 and NaBrO3 (Fig. 3c and d). As shown in Fig. 3, the currents of the last two reduction peaks increased, indicating that the reduced species of 1 have electrocatalytic activity for H2O2 and NaBrO3 reduction. Also, the third reduced species of compound 1 exhibits better electrocatalytic activity, that is, the catalytic activity is enhanced with the increasing extent of POM reduction.
Experimental section
Synthesis
A mixture of Htrz (0.08 g, 1.16 mmol), AgNO3 (0.07 g, 0.412 mmol), H5PMo10V2O40 (0.5 g, 0.288 mmol) and 4,4-bpy (0.0285 g, 0.183 mmol) was added to 10.0 mL of distilled water. After stirring for 2 h, the pH of the mixture was adjusted to 7.3 with saturated Na2CO3 solution. Then, the mixture was transferred to a 25 mL Teflon-lined autoclave, which was heated at 180 °C for 3 days and then cooled down to room temperature at a rate of 5 °C h−1. Yellow brown stripe crystals were isolated with a high yield of 58% (based on Mo) and washed for 5 times with distilled water. Anal. calcd for C28H24N16PMo12Ag9O41: C 9.91, N 6.06, Ag 28.61, P 0.91, Mo 33.92%; found: C 9.72, N 6.17, Ag 29.06, P 0.83, Mo 34.67%.
Preparation of 1-CPE
The 1-modified carbon paste electrode (1-CPE) was fabricated as follows: graphite powder (50 mg) and 1 (50 mg) were mixed and grounded together by an agate mortar to achieve a uniform mixture. Then 0.6 mL of nujol was added to the mixture, which was further stirred for about 1 hour. The homogenized mixture was packed into a glass tube with a 2 mm inner diameter, and the tube surface was wiped with paper. Electrical contact was established with a copper rod through the back of the electrode.
Instrumentation
Elemental analyses for Mo, P and Ag were determined by a Leaman inductively coupled plasma (ICP) spectrometer, and C and N were carried out with a Perkin-Elmer 2400 CHN Elemental analyzer. IR spectrum was obtained in the range 400–4000 cm−1 on an Alpha Centaurt FT/IR spectrophotometer using KBr pellets. TG analyses were recorded on a Perkin-Elmer TGA7 instrument in flowing N2 with a heating rate of 10 °C min−1. Powder X-ray diffraction (XRD) data was recorded by using a Rigaku D/max-2550 diffractometer with Cu-Kα radiation. GC-MS was collected by Agilent 5975-6890N. The HPLC analysis was obtained by using a Shimadzu LC-15C instrument.
X-ray crystallographic study
A single crystal of compound 1 was glued on the top of a glass fiber, and the crystallographic data was collected at room temperature on a Bruker Smart Apex CCD diffractometer with Mo-Kα monochromated radiation (λ = 0.71073 Å). The structure of compound 1 was solved by the direct method and refined by the full-matrix least-squares method on F2 using the SHELXTL-97 crystallographic software package.16 In the refinement, the restraint command ‘isor’ was employed to restrain several O and N atoms so as to avoid the ADP and NPD problems in the crystal data. Such refinement led to the restraint value of 42. The hydrogen atoms attached to organic ligands were fixed in calculated positions. The crystal data and structure refinements of compound 1 were summarized in Table 2.†
Table 2 Crystal data and structure refinement for 1
| Compound 1 |
| R1 = Σ||F0| − |Fc||/Σ|F0|; wR2 = Σ[w(F02 − Fc2)2]/Σ[w(F02)2]1/2. |
| Formula |
C28H24Ag9Mo12N16O41P |
| T (K) |
296(2) |
| Fw |
3393.71 |
| Crystal system |
Tetragonal |
| Space group |
I4/mcm |
| a (Å) |
18.0182(7) |
| b (Å) |
18.0182(7) |
| c (Å) |
19.6454(15) |
| V (Å3) |
6378.0(6) |
| Z |
4.00 |
| μ (mm−1) |
5.095 |
| λ (Å) |
0.71073 |
| R1, wR2 [I > 2σ(I)]a |
0.0594, 0.1511 |
| R1, wR2 (all data) |
0.0725, 0.1612 |
Conclusions
In conclusion, a new polyrotaxane structure constructed from the purely inorganic 1D chain and the 2D metal–organic layers has been synthesized by the hydrothermal method with a high yield. Compound 1 represents the first polyrotaxane compound consisting of the purely inorganic 1D POM chain and Ag-Htrz-4,4′-bpy networks. The Ag-Htrz-4,4′-bpy network was composed of two organic ligands, where the Ag+ ions was connected by them into the square Ag4O(Htrz)4 building unit and the liner Ag2(4,4-bpy) linking unit. It is noteworthy that the ratio of the mixed ligands played a decisive role in the synthesis of the title compound. This may suggest an efficient strategy for constructing the POM-based organic–inorganic hybrid materials. Furthermore, compound 1 exhibited efficient catalytic activities for oxidation of desulfurization, and electrocatalytic activity for reduction of H2O2 and BrO3−.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 21301020), Science and Technology Development Project Foundation of Jilin Province (20130522126JH/20150520001JH), Open Subject Foundation of Key Laboratory of Polyoxometalate Science of Ministry of Education, the Science and Technology Research Foundation of the Thirteenth Five Years of Jilin Educational Committee.
Notes and references
-
(a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature., 2003, 423, 705 CrossRef CAS PubMed;
(b) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS;
(c) B. H. Ye, M. L. Tong and X. M. Chen, Coord. Chem. Rev., 2005, 249, 545 CrossRef CAS;
(d) W. M. Xuan, C. F. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677 RSC;
(e) T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
-
(a) M. T. Pope, Heteropoly and IsopolyOxometalates, Springer Verlag, Berlin, 1983 Search PubMed;
(b) U. Kortz, A. Müller, J. Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315 CrossRef CAS;
(c) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736 CrossRef CAS PubMed.
-
(a) S. T. Zheng, J. Zhang, X. X. Li, W. H. Fang and G. Y. Yang, J. Am. Chem. Soc., 2010, 132, 15102 CrossRef CAS PubMed;
(b) X. J. Dui, W. B. Yang, X. Y. Wu, X. F. Kuang, J. Z. Liao, R. M. Yu and C. Z. Lu, Dalton Trans., 2015, 44, 9496 RSC;
(c) S. B. Li, H. Y. Ma, H. J. Pang and L. Zhang, Cryst. Growth Des., 2014, 14, 4450 CrossRef CAS;
(d) H. L. Wu, Z. M. Zhang, Y. G. Li, X. L. Wang and E. B. Wang, CrystEngComm, 2015, 17, 6261 RSC;
(e) Z. Zhang, J. Liu, E. Wang, C. Qin, Y. Li, Y. Qi and X. Wang, Dalton Trans., 2008, 463 RSC.
-
(a) D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Chem. Soc. Rev., 2014, 43, 4615 RSC;
(b) R. Li, X. Q. Ren, J. S. Zhao, X. Feng, X. Jiang, X. X. Fan, Z. G. Lin, X. G. Li, C. W. Hu and B. Wang, J. Mater. Chem. A, 2014, 2, 2168 RSC;
(c) X. T. Rao, T. Song, J. K. Gao, Y. J. Cui, Y. Yang, C. D. Wu, B. L. Chen and G. D. Qian, J. Am. Chem. Soc., 2013, 135, 15559 CrossRef CAS PubMed;
(d) C. Y. Duan, M. L. Wei, D. Guo, C. He and Q. J. Meng, J. Am. Chem. Soc., 2010, 132, 3321 CrossRef CAS PubMed.
-
(a) J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839 CrossRef CAS PubMed;
(b) L. H. Wee, F. Bonino, C. Lamberti, S. Bordiga and J. A. Martens, Green Chem., 2014, 16, 1351 RSC;
(c) W. Salomon, C. Roch-Marchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972 RSC;
(d) F. J. Ma, S. X. Liu, C. Y. Sun, D. D. Liang, G. J. Ren, F. Wei, Y. G. Chen and Z. M. Su, J. Am. Chem. Soc., 2011, 133, 4178 CrossRef CAS PubMed.
-
(a) Z. M. Zhang, T. Zhang, C. Wang, Z. K. Lin, L. S. Long and W. B. Lin, J. Am. Chem. Soc., 2015, 137, 3197 CrossRef CAS PubMed;
(b) A. X. Yan, S. Yao, Y. G. Li, Z. M. Zhang, Y. Lu, W. L. Chen and E. B. Wang, Chem.–Eur. J., 2014, 20, 6927 CrossRef CAS PubMed.
-
(a) L. M. R. Albelo, A. R. Ruiz-Salvador, A. Sampieri, D. W. Lewis, A. Gómez, B. Nohra, P. Mialane, J. Marrot, F. Sécheresse, C. Mellot-Draznieks, R. N. Biboum, B. Keita, L. Nadjo and A. Dolbecq, J. Am. Chem. Soc., 2009, 131, 16078 CrossRef PubMed;
(b) X. L. Wang, H. L. Hu, G. C. Liu, H. Y. Lin and A. X. Tian, Chem. Commun., 2010, 46, 6485 RSC;
(c) J. Q. Shen, Y. Zhang, Z. M. Zhang, Y. G. Li, Y. Q. Gao and E. B. Wang, Chem. Commun., 2014, 50, 6017 RSC;
(d) S. Yao, J. H. Yan, H. Duan, Z. M. Zhang, Y. G. Li, X. B. Han, J. Q. Shen, H. Fu and E. B. Wang, Eur. J. Inorg. Chem., 2013, 4770 CrossRef CAS;
(e) S. Z. Li, D. D. Zhang, Y. Y. Guo, P. T. Ma, J. W. Zhao, J. P. Wang and J. Y. Niu, Eur. J. Inorg. Chem., 2011, 5397 CrossRef CAS.
-
(a) S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959 CrossRef CAS PubMed;
(b) K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
-
(a) X. L. Wang, C. Qin, E. B. Wang and Z. M. Su, Chem. Commun., 2007, 4245 RSC;
(b) H. Y. Zang, Y. Q. Lan, G. S. Yang, X. L. Wang, K. Z. Shao, G. J. Xu and Z. M. Su, CrystEngComm, 2010, 12, 434 RSC;
(c) X. L. Wang, C. Xu, H. Y. Lin, G. C. Liu, S. Yang, Q. Gao and A. X. Tian, CrystEngComm, 2012, 14, 5836 RSC.
-
(a) D. Liu, Y. Lu, H. Q. Tan, W. L. Chen, Z. M. Zhang, Y. G. Li and E. B. Wang, Chem. Commun., 2013, 49, 3673 RSC;
(b) A. Nisar, J. Zhuang and X. Wang, Adv. Mater., 2011, 23, 1130 CrossRef CAS PubMed.
-
(a) G. F. Hou, L. H. Bi, B. Li and L. X. Wu, Inorg. Chem., 2010, 49, 6474 CrossRef CAS PubMed;
(b) X. L. Wang, Y. F. Bi, B. K. Chen, H. Y. Lin and G. C. Liu, Inorg. Chem., 2008, 47, 2442 CrossRef CAS PubMed;
(c) S. B. Li, W. Zhu, H. Y. Ma, H. J. Pang, H. Liu and T. T. Yu, RSC Adv., 2013, 3, 9770 RSC.
-
(a) R. N. Adams, Anal. Chem., 1958, 30, 1576 CrossRef CAS;
(b) L. Gorton, Electroanalysis, 1995, 7, 23 CrossRef CAS.
-
(a) K. Unoura and N. Tanaka, Inorg. Chem., 1983, 22, 2963 CrossRef CAS;
(b) A. X. Tian, Y. L. Ning, J. Ying, G. C. Liu, X. Hou, T. J. Li and X. L. Wang, CrystEngComm, 2015, 17, 5569 RSC.
-
(a) Y. Liu, M. K. Wang, F. Zhao, Z. A. Xu and S. J. Dong, Biosens. Bioelectron., 2005, 21, 337 CrossRef PubMed;
(b) L. Zhang and S. J. Dong, J. Electroanal. Chem., 2004, 568, 184 Search PubMed.
-
(a) X. L. Wang, Z. H. Kang, E. B. Wang and C. W. Hu, J. Electroanal. Chem., 2002, 523, 142 CrossRef CAS;
(b) B. X. Dong, L. Chen, S. Y. Zhang, J. Ge, L. Song, H. Tian, Y. L. Teng and W. L. Liu, Dalton Trans., 2015, 44, 1435 RSC;
(c) L. Cheng, X. M. Zhang, X. D. Xi and S. J. Dong, J. Electroanal. Chem., 1996, 407, 97 CrossRef.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, IR, HPLC, MS, PXRD and CIF file. CCDC 1434784. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra23830g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 














