
Unsaturated polyfluoroalkyl ketones in the synthesis of nitrogen-bearing heterocycles
A. Yu. Rulev
* and
A. R. Romanov
A. E. Favorsky Institute of Chemistry, Siberian Division of the Russian Academy of Sciences, Irkutsk 664033, Russia. E-mail: rulev@irioch.irk.ru; Fax: +7 3952 419346; Tel: +7 3952 511 429
First published on 18th December 2015
Abstract
This review focuses on the recent achievements and new developments in the synthesis of polyfluorinated aza-heterocycles based on fluorine-bearing enones and ynones published in the last decade.
 A. Yu. Rulev | Alexander Yu. Rulev studied chemistry at the Irkutsk State University and obtained his first post-graduate scientific degree (PhD equivalent) in 1988. He then worked as a researcher in the laboratory of organoelement compounds of the Institute of Organic Chemistry headed by academician M. G. Voronkov. In 1995, he received a NATO Fellowship to conduct research in the Laboratoire des Fonction Azotées et Oxygénées Complexes de l'Université de Rouen (France) with Prof. P. Duhamel and Dr L. Duhamel. In 2002 he obtained a scientific degree of Doctor of Sciences (Chemistry). His research focuses on the chemistry of carbonyl-bearing captodative and push–pull aminoalkenes, and aza-Michael reactions. He has over 70 papers and two popular science books. |
 A. R. Romanov | Alexey R. Romanov, born in 1989 in Irkutsk, studied chemistry at the Irkutsk State University, gaining his diploma in 2012. Now he is doing research at the A. E. Favorsky Institute of Chemistry under the supervision of Dr A. Rulev and going to present his thesis on the synthesis of polyfluorinated aza-heterocycles based on unsaturated ketones and binucleophiles. |
1. Introduction
The interest in the development of versatile and effective approaches to fluorine-containing organic compounds is due to the search for new materials possessing unique characteristics. It is well known that incorporation of fluorine atom(s) and/or fluorinated functional group(s) into molecules of organic compounds often furnishes derivatives bearing remarkable chemical, physical and biological properties. Therefore it is not surprising that this modification is often used in materials science and agricultural, biological and pharmaceutical chemistry.1,2 According to reported data, fluorine is incorporated into 10–15% of drugs synthesized during last half a century, and their number increased in the beginning of 21st century.3–12 The molecules of more than 200 currently used drugs contain at least one fluorine atom.13 In the past decades polyfluoroalkylated heterocyclic compounds are paid a great attention due to their use as agrochemicals and medicine. Among them, nitrogen-bearing heterocycles are of particular interest: they are recognized as privileged pharmacophores.14–16 The currently used methods of their synthesis can be divided into two groups. The first approach presupposes the incorporation of polyfluoroalkyl moiety into existing heterocyclic core; the second one is based on the use of fluorine-containing synthones. The known methods of direct fluorination do not always allow the introduction of fluorine atom at the required position of molecule. Moreover, the hazard and toxicity of fluorinating reagents as well as the use of expensive equipment and tedious procedures significantly narrow the area of its application.17 The use of fluorine-containing building blocks is more convenient approach to target compounds.18,19 One-pot synthesis methodology intensively developing in the past decades provides the reducing of the number of reaction steps and amount of costs and waste products.20,21 The reaction of unsaturated polyfluoroalkylketones with bidentate nucleophiles is considered nowadays as an effective approach to fluorinated heterocyclic compounds (Scheme 1).2,22–24 Nucleophilic attack can be directed to carbonyl carbon as well as Cβ of triple or double bond (in the latter case ring assembly initiated by aza-Michael reaction (aza-MIRC reactions) is implied).25 The size and type of forming heterocycle are defined by both nature of reagents and reaction conditions. Various synthetic strategies have been proposed to assembly nitrogen heterocycles but the development of new efficient approaches to these derivatives is an ongoing challenge of the modern organic synthesis. | ||
| Scheme 1 | ||
This review focuses on recent advances in the synthesis of fluorine-bearing aza-heterocycles from α,β-unsaturated polyfluoroalkylketones (mainly, CF3-ynones and functionalized CF3-enones) and bidentate nucleophiles, covering the data reported over the last seven years after the publication of Druzhinin et al.22 The classification of this review is based on the type of assembled heterocycle.
2. Synthesis of five-membered heterocycles
2.1. Pyrroles
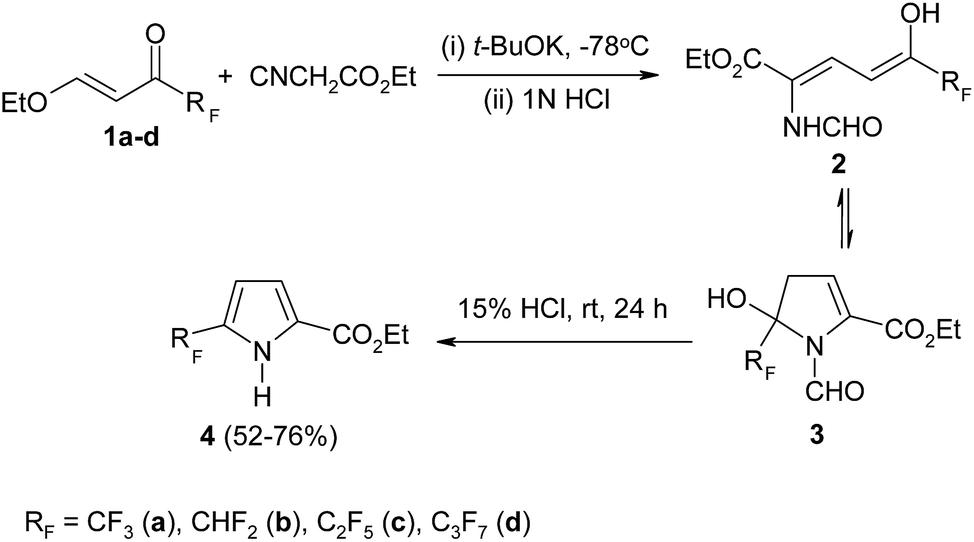
In spite of the fact that a number of approaches to CF3-pyrroles was developed, the elaboration of novel methods of their preparation was attracting the attention in the past decade. Nowadays, the trifluoromethyl(β-alkoxyvinyl)ketones are considered as valuable building blocks for construction of wide type of heterocyclic systems. It is not surprising because these substrates are typical push–pull olefins having a highly polarized double carbon–carbon bond. That is why they are very active Michael-acceptors in reactions with nucleophiles.Recently, fluorinated β-alkoxyenones 1 have been used as initial substrates in the original method of pyrrole core assembly (Scheme 2).26
 | ||
| Scheme 2 | ||
As should be expected, the 1,2-addition of ethylisocyanate to enones 1 is found to be a minor process while the main reaction starts with nucleophilic attack on Cβ-atom leading to product 2. The latter exists in solution as a mixture of tautomers including cyclic form 3, which is direct precursor to corresponding pyrrole 4. Under mild conditions the derivatives 3 easily eliminate the molecule of formic acid and transform into polyfluoroalkylated pyrroles 4 which are isolated in good yields.
2.2. Pyrazoles
Trifluoromethylpyrazoles are generally obtained by cyclocondensation of 1,3-diketones with hydrazines. However, despite the high yields of target heterocycles this method for synthesis of CF3-containing pyrazoles has a substantial drawback: reactions of monosubstituted hydrazines with unsymmetrical ketones often lead to inseparable mixture of regioisomers.27 The use of synthetic equivalents of 1,3-diketones – acetylenic or β-functionally substituted fluorine-containing ketones – is more attractive. Indeed, for the first time CF3-pyrazoles were easily obtained from trifluoromethylated ynone 5a and hydrazine.28 Monosubstituted hydrazines a priori can give the mixture of isomeric pyrazoles. The reaction direction depends on its conditions and the structure of initial compounds. For instance, the reaction of phenylhydrazine with CF3-ynone 5 in aqueous acetonitrile at room temperature results in formation of 3-CF3-pyrazole 6 exclusively.29 The replacement of CF3-moiety by CCl3-group in initial ketone affords its 5-substituted analogue 7 in good yield (Scheme 3).30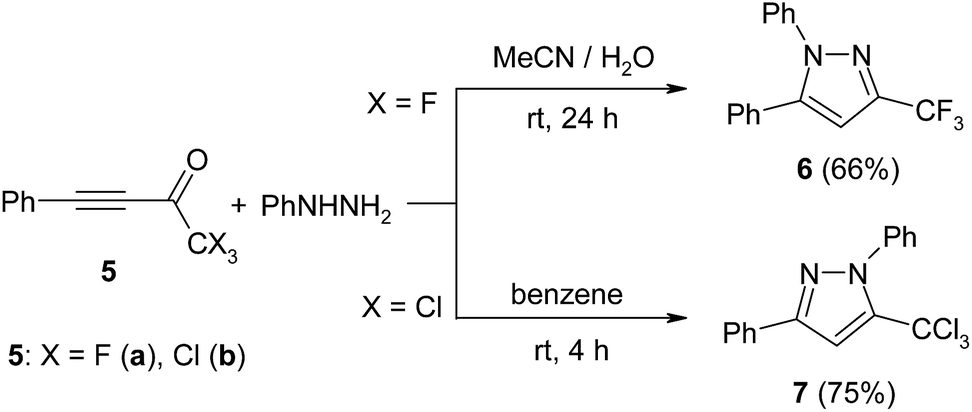 | ||
| Scheme 3 | ||
Selective synthesis of 5-substituted pyrazoles 9 is based on reaction of polyfluoroalkyl(alkynyl)ketones 8 with substituted hydrazines catalyzed by Lewis acids (Scheme 4).31
 | ||
| Scheme 4 | ||
Very recently, a remarkable achievement in regio-switchable synthesis of trifluoromethylated pyrazoles has been reported.32 It was shown that CF3-ynones 10 react with monosubstituted aryl- and alkylhydrazines to give preferentially either 3- or 5-trifluoromethylpyrazoles 11 and 12 depending on reaction conditions (Scheme 5). When the reaction was carried out in highly polar aprotic solvent such as DMF and (especially) DMSO in the absence of any catalyst, the formation of pyrazoles 11 was observed. In contrast, the isomeric pyrazoles 12 were obtained with high selectivity in the presence of catalyst and in low polarity aprotic solvent (such as CH2Cl2). According to proposed mechanism, the assembly of 5-CF3-pyrazoles 12 is initiated by aza-Michael addition of hydrazine derivative to ynone 10 followed by cyclization and dehydration to yield target heterocycle. In contrast, in the presence of Lewis acid (Cu(OAc)2) the reaction starts with the generation of acetylenic hydrazone which transforms into pyrazole 11.
 | ||
| Scheme 5 | ||
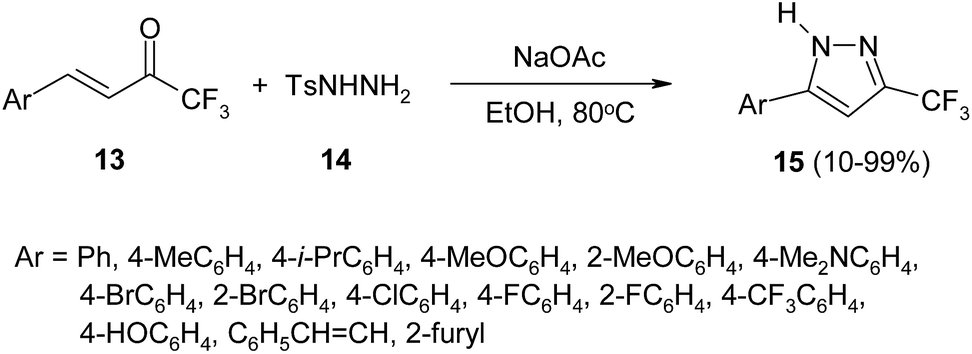
Trifluoromethylenones can also be used as starting materials for the preparation of CF3-pyrazoles. Thus, a simple procedure of the regioselective synthesis 3-trifluoromethylpyrazole 15 from enones 13 and tosylhydrazine 14 has been described.33 The reaction proceeds through usual cyclization followed by 1,5-H shift transformation leading to pyrazoles 15 in moderate to high yield (Scheme 6).
 | ||
| Scheme 6 | ||
Of late years, enormous number of articles devoted to synthesis of pyrazoles from functionally substituted CF3-enones (mainly β-alkoxy- and β-aminoenones) has been published. The main efforts of synthetic chemists are concentrated on expansion of area of reagents involved as well as development of “green” methods of selective assembly of pyrazole ring.
Polyfluoroalkylated β-alkoxy-34–36 and β-aminovinylketones37 as well as chromones38 react with hydrazine hydrate to give required heterocycles. Thus, pyrazole 17 was obtained in high yield after holding of equimolar mixture of β-ethoxyenone 16 with hydrazine in acetic acid at room temperature (Scheme 7).36
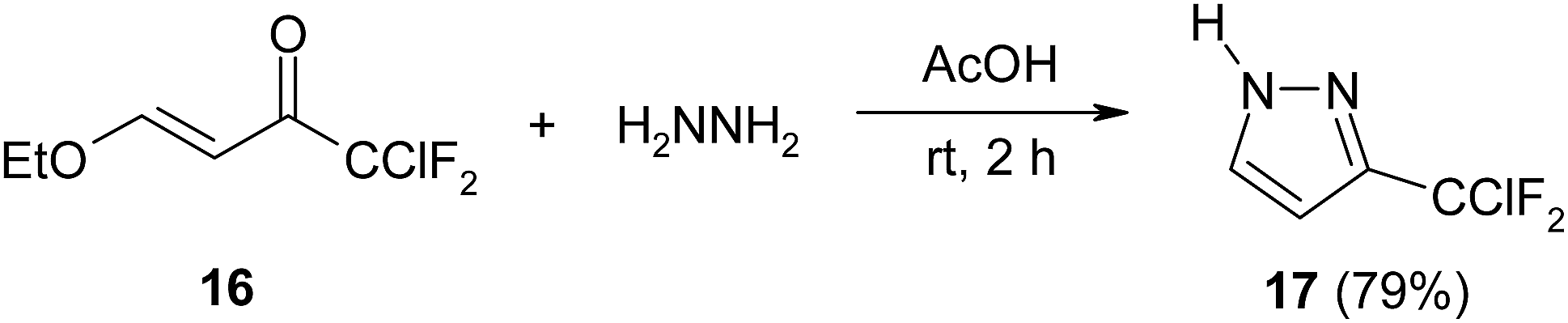 | ||
| Scheme 7 | ||
Depending on reaction conditions, (methoxyalkenyl)enone 18 reacts with hydrazine hydrate to form either mono- or bis-pyrazoles 19 and 20 (Scheme 8).34
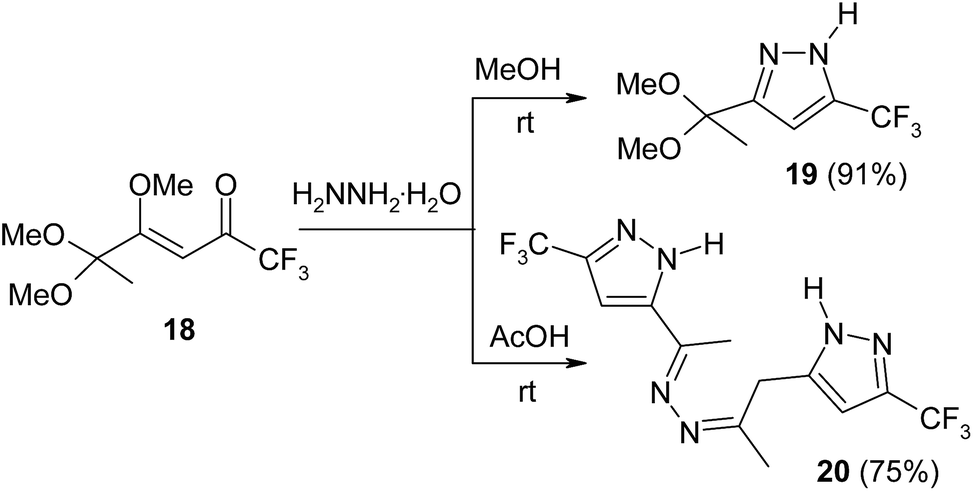 | ||
| Scheme 8 | ||
Recently, Nenajdenko et al. described the synthesis of bromine-bearing CF3-pyrazole 22 from α,β-dibromoenone 21 and hydrazine hydrate.39 However, its analogue 23 containing no bromine was also formed. The authors hypothesized that the latter heterocycle is the result of halophilic attack of hydrazine to α-bromine atom of initial ketone. The reaction proceeds at reflux for 10–12 h leading to the mixture of brominated and non-brominated pyrazoles which were not separated (Scheme 9). Despite the good yields in some solvents, this method will be hardly used for the synthesis of such type of functionalized pyrazoles.
 | ||
| Scheme 9 | ||
The most intriguing problem in pyrazole synthesis from β-functionally substituted (polyfluoroalkyl)enones and monosubstituted hydrazines is the selectivity of addition reaction initiating pyrazole core assembly. Analysis of reported data indicates that cyclocondensation of these enones with alkyl- and arylhydrazines is much more selective process than the same procedure for non-fluorinated derivatives.
Generally, the major product (in some cases the sole product) in this reaction is 5-trifluoromethyl-1H-pyrazole. The formation of isomeric 3-trifluoromethyl-1H-pyrazoles is much more rare process. In spite of the abundance of material, it is quite difficult to predict accurately the structure of the major product. Authors40 supposed that the reaction direction depends more on reactivity of substrate and less on the nature of substituents in hydrazine. However, this supposition is not always confirmed practically. Often, the relatively trivial changes in the structure of initial reagents and reaction conditions (method of activation, temperature, solvent, the presence of additional base) can lead to different types of products.
The examples combined in Table 1 show that pyrazoles are mostly obtained with refluxing of initial reagents in alcohols. However, the process under these conditions is quite long and not always effective. Recently, the successful efforts of synthesis of pyrazoles were applied in ionic liquids,40–42 combined with microwave irradiation41–46 or in supercritical CO2 used as organic solvent substitute.47
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Enone | R | Conditions | Product, yield (%) | Ref. | ||
| R1 | R2 | R3 | |||||
a Ratio B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D varies from 100:0 to 88:12.b Ratio B:D varies from 15:85 to 57:43; when NaOH was used instead of pyridine, pyrazole D was obtained only.c Ratio B:D varies from 20:80 to 80:20.d Ratio B:D is 35:65.e In some cases a mixture of isomeric pyrazoles B and D (ratio varies from 1:1 to 1:10) was obtained.f Ratio B:D varies from 50:50 to 80:20. :D varies from 100:0 to 88:12.b Ratio B:D varies from 15:85 to 57:43; when NaOH was used instead of pyridine, pyrazole D was obtained only.c Ratio B:D varies from 20:80 to 80:20.d Ratio B:D is 35:65.e In some cases a mixture of isomeric pyrazoles B and D (ratio varies from 1:1 to 1:10) was obtained.f Ratio B:D varies from 50:50 to 80:20. |
|||||||
| 1 | H, Ph, 4-XC6H4 (X = Me, MeO, F), 2-furyl | H, Me | MeO, EtO | Ph | MeCN, rfx, 24 h | B + D (78–95)a | 48 |
| 2 | H, Me, Ph | H | MeO, EtO | t-Bu | [BMIM][BF4], Py, 78 °C, 15 h or EtOH, Py, 78 °C, 15 h | B + D (70–93)b | 40 |
| 3 | H, Me, Ph | H | MeO | Ph | sc-CO2, 60–120 bar, 65–80 °C, 45 min | D (45–89) | 47 |
| 4 | H, Me, Ph | H | MeO, EtO | Ph, (CH2)2OH | Solvent free, MW, 1–12 min | B + C + D (70–95)c | 43 |
| 5 | Cl, Br | H | Cl, Br | Et, Bn | EtOH, Et3N, rfx, 3 h | B (49–74) | 49 |
| 6 | MeS | H | MeS | Ph | EtOH, rfx, 2 h | B + D (≤20)d | 50 |
| 7 | H, Me, Bu, i-Bu, Ph, 4-XC6H4 (X = Me, F, Cl, Br, I), 2-furyl | H | MeO, EtO | Ph | [BMIM][BF4], 25–150 °C, 1–3 h or [BMIM][BF4], MW, 6 min | D (56–96)e | 42 |
| 8 | Me, Et, Pr, i-Pr, Ph, 4-FC6H4, 4-BrC6H4 | H | MeO | C6F5 | Solvent free, MW, 6 min or [BMIM][BF4], 80 °C, 1 h | C (75–94) | 41 |
| C (43–82) | |||||||
| 9 | Me | CO2But | MeNH | Me, Et, (CH2)2OH | MeOH, −20 °C → rt | B + D (63–95)f | 51 |
| 10 | H | CO2Et | EtO | 4-NCC6H4 | EtOH, rt, 16 h | D (77) | 52 |
| 11 | H | CO2Et | EtO | 4-O2NC6H4 | EtOH, rfx, 16 h or MW, 2 min | D (82) | 44 |
| 12 | H | CO2Et | EtO | Thiazolyl | EtOH, rfx, 30–40 min | D (62–88) | 53 |
| 13 | H, CF3 | H, CO2Et | EtO | 4-O2NC6H4, 4-BrC6H4 | MeOH, EtOH, PrOH, DMF, MW, 1.5–15 min | D (81–98) | 45 |
| 14 | (MeO)2CHCH2 | H | MeO | C6F5, 2-furyl | EtOH, rfx, 4–20 h | C (90–97) | 54 |
| 15 | (MeO)2CHCH2 | H | MeO | Me, t-Bu, Ph | MeOH, rfx, 20 h | D (89–98) | 55 |
| 16 | MeO2C(CH2)2 | H | MeO | Ph | MeOH, rfx, 8 h | D (94) | 56 |
| 17 | Me2S(O)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) |
H | EtO | Ph | EtOH, rfx, 16 h | D (78) | 57 |
| 18 | H, Me, Et, Pr, i-Pr, Bu, i-Bu, Ph | H | MeO, EtO | Ph | Toluene, MW, 200 W, 3 min | C (80–91) | 46 |
| Toluene, MW, 300 W, 10 min | D (83–90) | ||||||
| 19 | H | (CH2)nO, n = 1, 2 | C6F5, Ph | EtOH, rfx, 20 h | C (60–72) | 58 | |
| D (89–93) | |||||||

The application of microwave irradiation provides not only decreasing of the reaction time (from several hours to several minutes compared to conventional heating) but yield increase of target heterocycles as well (including nitrophenyl-substituted hydrazines) (Table 1, entries 8, 11, 13). The type of heterocycle also depends on the irradiation rate: dihydropyrazoles are formed at 200 W whereas at 300 W the formation of pyrazoles was only observed (Table 1, entry 18).
The formation of pyrazoles successfully occurs in ionic liquids. Among them 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4] showed the highest catalytic activity (Table 1, entries 2, 7, 8). Synergic effect of ionic liquid and microwave irradiation provided shorter reaction time (up to 6 min) without decreasing yield of target heterocycles (Table 1, entry 7).
Pyrazoles were recently prepared under solvent free conditions.43,47 This approach attractive from both economical and ecological point of view turned out to be very effective. For instance, mono-substituted β-methoxy-CF3-enone and its analogues, bearing alkyl (Me) or aryl (Ph) substituents, react with phenylhydrazine, heated in supercritical carbon dioxide, to form 5-trifluoromethyl-1H-pyrazoles (Table 1, entry 3). The apparent merits of developed procedure are high ecological compatibility and facility of product isolation.
Original two-step method of selective assembly of 3-fluoromethylated pyrazoles 26 was proposed by Zanatta and coworkers (Scheme 10).48 Using hydrazones 24, prepared from phenylhydrazine and benzaldehyde and its derivatives, the authors obtained β-aminoenones 25, which undergo intramolecular cyclization into target pyrazoles 26 under acidic conditions. The selective formation of 3-CF3-pyrazoles makes this method more attractive compared to classic cyclocondensation reaction.
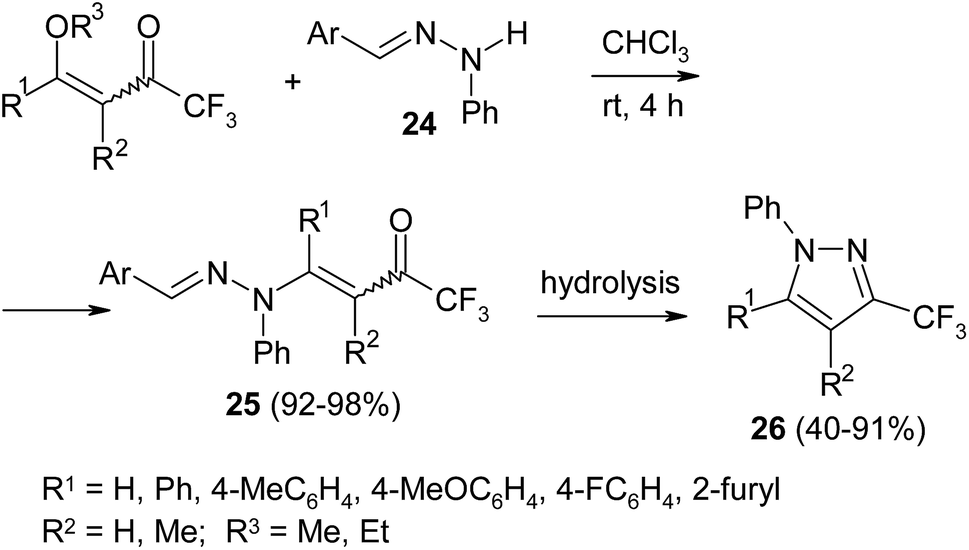 | ||
| Scheme 10 | ||
A priori, 1,3-diaminoguanidine 27 can react with β-methoxyalkenyl(trifluoromethyl)ketones as N–C–N or N–N binucleophile giving either pyrimidines or pyrazoles correspondingly. However, the latter case only is implemented practically: bis-pyrazoles 28 are smoothly prepared after 4–5 hours of refluxing in alcohol (Scheme 11).59
 | ||
| Scheme 11 | ||
Substituted hydrazides were also used as nucleophiles in the synthesis of pyrazoles. Compared to hydrazines, their reaction with β-alkoxyalkenyl(polyhaloalkyl)ketones occurs more selectively giving exclusively dihydropyrazoles 29 (Scheme 12). In almost all cases the required heterocycles were isolated in moderate to high yields after long refluxing of initial reagents in alcohol.60–69 As it was observed for hydrazines, the use of ionic liquids70 and microwave activation in the presence71 or absence72,73 of solvent shortened reaction time as well. The selectivity of reactions with hydrazides under these conditions was not affected at all.
 | ||
| Scheme 12 | ||
Interestingly, reactions of hydrazides with alkoxyenones and 1,3-dicarbonyl compounds proceeds in different ways. Thus, when CF3-enones were treated with hydrazide of cyanoacetic acid the dihydropyrazoles were obtained whereas 1,3-diketones furnished the pyridine derivatives.74
3-CF3-pyrazoles 30 were obtained in high yield from the reaction of β-alkoxyalkenyl(trifluoromethyl)ketones with phenylsemicarbazide (Scheme 13).75 The key steps of the mechanism of this transformation are: aza-Michael reaction with participation of more nucleophilic nitrogen atom attached to phenyl group, water elimination, intramolecular cyclization, and subsequent aromatization of five-membered ring. Reaction proceeds in alcohol in the presence of catalytic amounts of sulfuric acid. The method proposed stands out for the conducting facility of multi-step reaction and exclusive formation of 3-CF3-pyrazoles (the ratio of isomeric pyrazoles in most cases is 100:0).
 | ||
| Scheme 13 | ||
2.3. Isoxazoles
Generally, isoxazoles are obtained by cyclocondensation of β-dicarbonyl derivatives, ynones, and β-alkoxyvinylketones with hydroxylamine. The former is generated in situ from its salts (as a rule, hydrochloride) by organic (pyridine, alkali metals alcoholates) or mineral (alkali metals hydroxides and carbonates) bases.76 The main problem in the synthesis of these heterocycles is control of regioselectivity. The analysis of reported data showed that selectivity of isoxazole formation depends on the structure of initial reagents, stability of forming cycle, and reaction conditions. Thus, Linderman in his pioneer work, devoted to the synthesis of trifluoromethylated five-membered N-heterocyclic compounds, mentioned the formation of complex mixture of isoxazole 31, dihydroisoxazole 32, and oxime 33 in reaction of CF3-ynone 5 with hydroxylamine (Scheme 14).28 In analogous reaction the CCl3-ynone was selectively transformed into 5-trichloromethyl-4,5-dihydroisoxazole 34 in 70% yield.30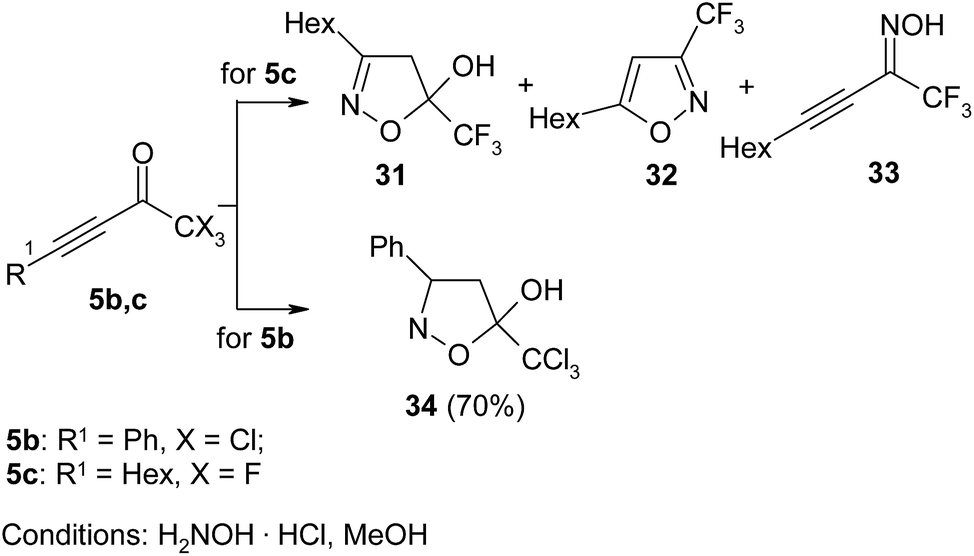 | ||
| Scheme 14 | ||
In the past decade new efficient synthetic routes to polyhaloalkyl-substituted isoxazoles were developed. Almost all of them are based on the reaction of push–pull alkoxyenones with hydroxylamine. The apparent advantage of using β-alkoxyalkenyl(polyhaloalkyl)ketones is almost complete independence of selectivity from any changes in reagent structure and reaction conditions. The presence of strong electron withdrawing group and, therefore, substantial difference in electrophilicity of β-olefinic carbon atom and CO moiety of substrate as well as a difference in nucleophilicity of two heteroatom centers of hydroxylamine provide an excellent selectivity of nucleophilic addition. At that, more nucleophilic nitrogen atom of hydroxylamine is added to Cβ atom of enone (aza-Michael reaction) whereas oxygen attacks the carbon of carbonyl group.
5-Hydroxy-4,5-dihydroisoxazoles are generally unstable and after elimination of water transform into isoxazoles. The stability of dihydroisoxazoles having strong electron withdrawing substituent at position 5 is so high that they can be easily isolated in a pure state by column chromatography. However, when 5-hydroxy-5-polyhaloalkyl-4,5-dihydroisoxazoles were treated with a strong dehydrating reagent (usually H2SO4), the corresponding isoxazoles were obtained. Some examples of the isoxazoles synthesis are combined in Table 2.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Enone | Conditions | Product, (yield, %) | Ref. | |||
| R1 | R2 | R3 | X | ||||
| a Transformation of CCl3 group into COOH occurs for CCl3 derivative. | |||||||
| 1 | (CH2)2CO2Me | H | MeO | F, Cl | MeOH, Py, rfx, 8 h | E (90–95%) | 56 |
| 2 | (CH2)2CO2Me | H | MeO | F, Cl | H2O, rfx, 12 h | F (68–75%)a | 56 |
| 3 | CH2CH(OMe)2 | H | MeO | F | Py, 45 °C, 24 h | E (73%) | 55 |
| 4 | Me2S(O)N |
H | EtO | F | MeOH, Py, 65 °C, 16 h | E (71%) | 57 |
| 5 | (EtO)2P(O)CH2 | H | O(CH2CH2)2N | F | MeOHaq., NaHCO3, rt, 3 h | E (27%) | 77 |
| 6 | Ar | H | MeO | F, Cl | (i) MeOH, Py, MW, 6 min | F (78–90%) | 79 |
| (ii) H2SO4 conc., MW, 10 min | |||||||
| 7 | (CH2)n; n = 5, 6, 10 | MeO | F | H2O, Py, 45 °C, 24 h | E (61–85%) | 82 | |
| 8 | Ar | H | MeO | F | MeOH, HCl, 70 °C, 48 h | F (67–80%) | 78 |
| 9 | MeS | H | MeS | F | MeOH, KOH, rfx, 4 h | E (54%) | 50 |

Besides β-alkoxyvinylketones, their morpholine-substituted analogue can be used in reaction with hydroxylamine (Table 2, entry 5). But in this case, however, the yield of required heterocycle is lower.77
Generally, 5-hydroxy-4,5-dihydroisoxazoles are successfully prepared from both CF3- and CCl3-enones (Table 2, entry 1). But under dehydratation conditions the CCl3-moiety often undergoes transformation onto carboxylic function (Table 2, entry 2). The new method of the isoxazoles synthesis under microwave irradiation not only reduces the reaction time and amount of waste products (in comparison with classical method78) but saves CCl3-group as well.79
In contrast with non-fluorinated analogues, 3-(polyfluoroacyl)chromones 35 easily react with hydroxylamine hydrochloride in methanol by 1,4-addition scheme, leading to annelated isoxazoles 36 (Scheme 15).80
 | ||
| Scheme 15 | ||
Finally, nitriloxides participate in 1,3-dipolar cycloaddition to β-ethoxyvinyl(trifluoromethyl)ketone to give bicyclic compounds 37 containing both isoxazole and dioxazole moieties (Scheme 16).81 Considering tendency of nitriloxide to dimerization, the authors generated the latter in situ from hydroxamyl chloride by basic reagents. The best results were obtained when reaction proceeded in benzene at room temperature in presence of NHCO3.
 | ||
| Scheme 16 | ||
3. Synthesis of six-membered heterocycles
3.1. Pyrimidines
Being a structural fragments of nucleic acids and drugs, pyrimidines attract particular attention of synthetic chemists. There are many methods of assembly of pyrimidine core, but the development of new and modification of already existing approaches remain the actual goal of modern organic chemistry. The emergence of new pharmaceutical drugs possessing broad spectrum of activities stimulates the search for novel reagents for the synthesis of pyrimidine derivatives.One of the common approaches to these heterocycles involves the cyclocondensation reaction of 1,3-dicarbonyl compounds and their equivalents with binucleophiles bearing N–C–N moiety. Since the beginning of nineties of 20th century β-alkoxyvinyl(perfluoroalkyl)ketones have been intensively used in reaction with amidines, urea and its derivatives (such as thiourea and guanidines).
The reaction with urea in common solvents proceeds, as a rule, under harsh conditions in presence of catalytic amounts of Lewis or Bronsted acids leading to target heterocycles in mild yields (mainly, 50–60%) after boiling for a long time (20–480 h) (Table 3, entries 2–6). In ionic liquids the reaction time is shorter (3–6 h) and yield is better (up to quantitative).83 Push–pull aminoenones react with thiourea to give pyrimidines in low yield (Table 3, entry 7).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Enone | Urea | Conditions | Yield, % | Ref. | |||
| R1 | R2 | R3 | RF | X | ||||
| 1 | Ph, 4-YC6H4 (Y = MeO, Me, F, Cl, Br, I), 2-thienyl, 2-naphthyl | H, Me | MeO | CF3 | O | [BMIM][BF4], HCl, 100 °C, 3–6 h | 70–97 | 83 |
| 2 | H | CO2Et | EtO | C3F7 | O | DMF, 80 °C, 6–8 d; AcOH, rfx, 14 d | 56 | 90 |
| 3 | H, Me | H, Me | MeO, EtO | CF3 | O | MeOH, HCl, rfx, 20 h | 61–90 | 91 |
| 4 | 2-Furyl, 2-thienyl | H | MeO | CF3 | O | i-PrOH, BF3·Et2O, 45–50 °C, 20 h | 48–52 | 92 |
| 5 | (CH2)2CO2Me | H | MeO | CF3 | O | MeOH, rfx, 20 h | 57–71 | 93 |
| 6 | (CH2)n; n = 5, 6, 10 | MeO | CF3 | O | i-PrOH, BF3·Et2O, rfx, 20 h | 50–58 | 82 | |
| 7 | (EtO)2P(O)CH2 | H | Morpholine | CF3 | S | MeOH, H2O, HCl, rt, 144 h | 21–23 | 77 |
| 8 | 2-Furyl, 2-thienyl | H | MeO | CF3 | NH | MeOH, NaOH, 50 °C, 1 h | 50–67 | 92 |

The reactions of β-alkoxyvinyl(polyhaloalkyl)ketones with methylthiopseudourea or 1,2-dimethylisothiourea result in formation of di- or tetrahydropyrimidines depending on reaction conditions and structure of initial enone.84,85 For instance, the treatment of trifluoro- 38a or trichloromethylketone 39a with 1,2-dimethylisothiourea generated from its salt by 1 M NaOH at room temperature furnishes the tetrahydropyrimidines 40a and 41a correspondingly (Scheme 17).84 When reaction mixture of enone 39a with the same nucleophile is heated, the elimination of CCl3 group occurs to form pyrimidinone 43a in almost quantitative yield. In contrast with ketones 38a and 39a, their methyl-substituted analogues 38b and 39b under the same conditions give dihydropyrimidines 42b and 43b correspondingly. Authors explained this result by steric interactions of methyl substituents of enone and nucleophile.
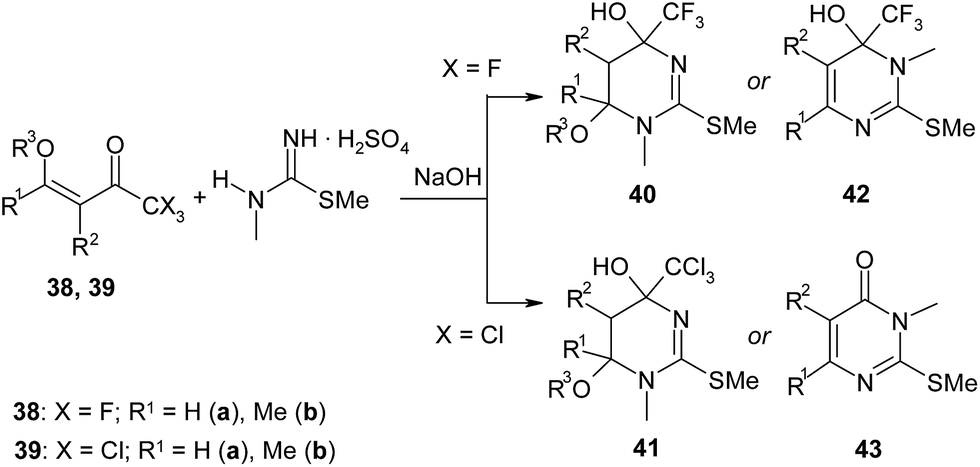 | ||
| Scheme 17 | ||
Non-substituted guanidine generated in situ from its salt by aqueous alkali solvent reacts with 4-(2-hetaryl)-4-methoxy-1,1,1-trifluoromethyl-3-buten-2-one giving pyrimidines in mild yield (Table 3, entry 8).
To study the chemoselectivity of cycloaddition the authors of articles55,57,86–89 used N-substituted guanidines as unsymmetrical binucleophiles bearing N–C–N moiety in reaction in reaction with β-alkoxyalkenyl(trifluoromethyl)ketones. Thus, CF3-pyrimidines 44 were obtained when N-acetylguanidine was treated with β-alkoxyenones in boiling acetonitrile or isopropanol (Scheme 18).55,57,86
 | ||
| Scheme 18 | ||
Interestingly, aminoguanidine reacts with β-methoxyalkenyl(trifluoromethyl)ketone 45 to give bicyclic compound 46 containing both five- and six-membered rings (Scheme 19).56 When heated in the presence of sulfuric acid the intermediate 46 dehydrated smoothly to give pyrazolylpyrimidine 47 in high yield. The unique role of trifluoromethyl group in assembly of derivative 47 is confirmed by the fact that efforts to obtain its CCl3-analogue under the same conditions failed.56 It should be noted that, in contrast to monoaminoguanidine, 1,3-diaminoguanidine reacts with β-methoxyalkenyl(trifluoromethyl)ketones exclusively as hydrazine leading to formation of bis-pyrazole derivatives (see chapter 2.2): pyrimidine core has not been formed in this case.59
 | ||
| Scheme 19 | ||
Besides urea derivatives, amidines can also be used as binucleophiles in preparation of pyrimidines.36,50,94–96 In the study of the reaction of arylamidines with 4-ethoxyvinyl(difluoro)ketone, the crucial role of solvent in successful assembly of pyrimidine core was shown (Scheme 20).94 As expected, the first step – aza-Michael addition – is promoted by protic solvents: the reaction proceeds smoothly in ethanol at temperature 5 °C after generating of amidine from its salt. The intermediate formed is quite stable: it remains almost unchanged during several hours at 35 °C. The next step – aromatization – is a limiting stage for the whole cascade of transformations. In alcohols this transformation proceeds slowly causing required heterocycles to be formed in mild yields. On the contrary, polar aprotic solvents (DMSO, DMF, DMAc) have an accelerating effect on this reaction shifting the equilibrium between base and amidine anion towards latter which results in rate increase of elimination of alkoxy-group and aromatization of cycle.
 | ||
| Scheme 20 | ||
Scientific teams from Russia and Brazil have obtained series of pyrimidine derivatives by reaction of polyfluoroalkylated β-alkoxyenones with aminosubstituted pyrazoles,97–101 triazoles,100,102–104 imidazoles101 and benzimidazoles.105,106 It was shown that results are strongly dependent on reaction conditions. Thus, Goryaeva et al. reported that aminopyrazoles as N,N-binucleophiles easily react with carbonyl compound 48 to form pyrimidinecarboxylates 49 with a good yield (Scheme 21).98 After long boiling in glacial acetic acid these derivatives undergo transformation into pyrazolopyrimidines 50. On the contrary, their isomers, pyrazolopyridines 51, were unexpectedly isolated when crude products were recrystallized from alcohol. According to the authors's assumption, this transformation proceeds via acyclic intermediate 52 which undergoes an intramolecular Michael addition. This type of recyclization proceeds peculiarly in the case of perfluoroalkylated pyrazolopyrimidines and is not observed for non-fluorinated analogues.
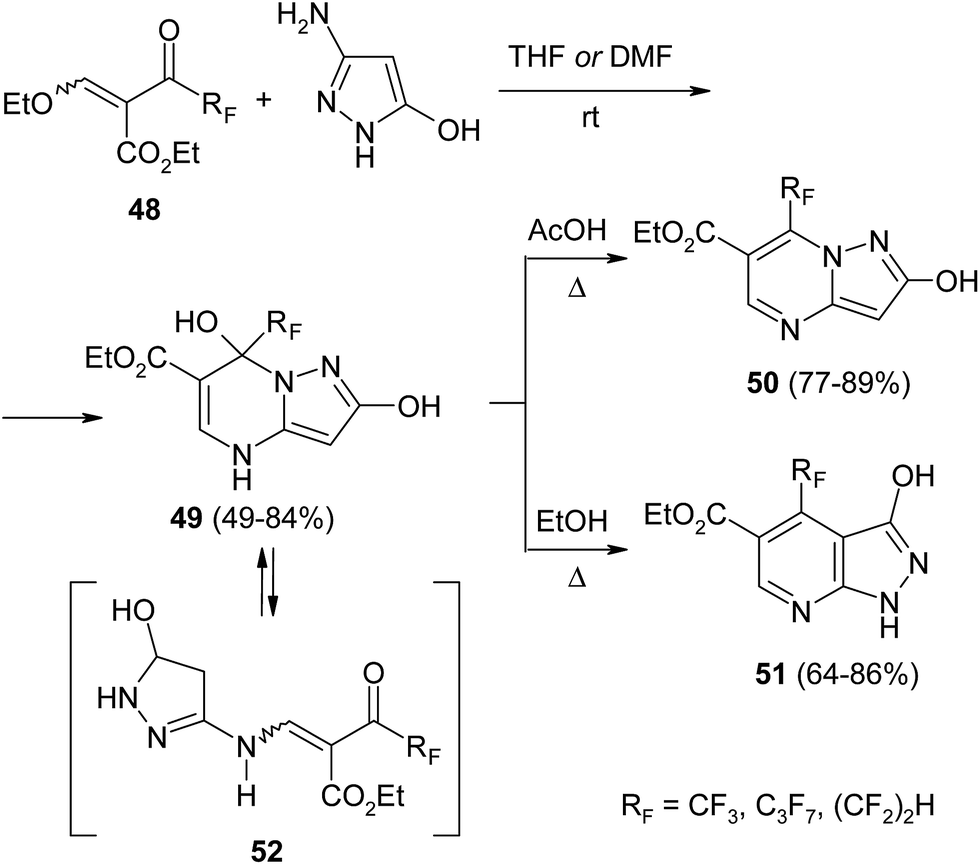 | ||
| Scheme 21 | ||
Aminopyrazoles and aminotriazoles react under mild conditions with CF3-enones to afford exclusively corresponding azolopyrimidines 53 (Scheme 22).100 The authors managed to register the key intermediate – pyrazolopyrimidinol – and monitor its gradual transformation into final reaction product.
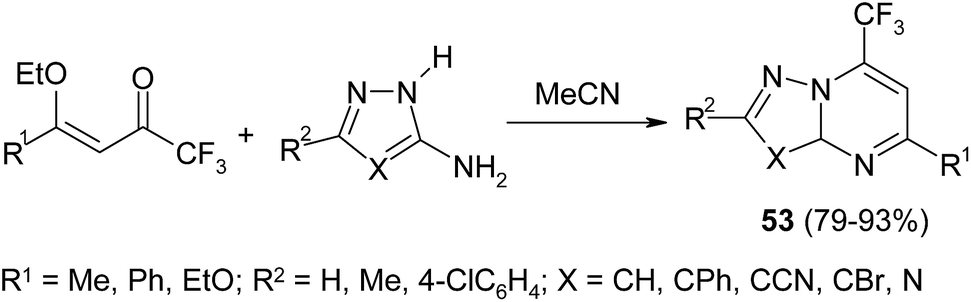 | ||
| Scheme 22 | ||
Recently ultrasound irradiation has been utilized to accelerate the assembly of triazolopyrimidines 55. Thus, conventional heating of the mixture of methoxyenones 54 and 5-amino-1,2,4-triazole in acetic acid affords the target heterocycles in 6 hours whereas ultrasound irradiation reduces reaction time up to 5–15 minutes (Scheme 23).99,102
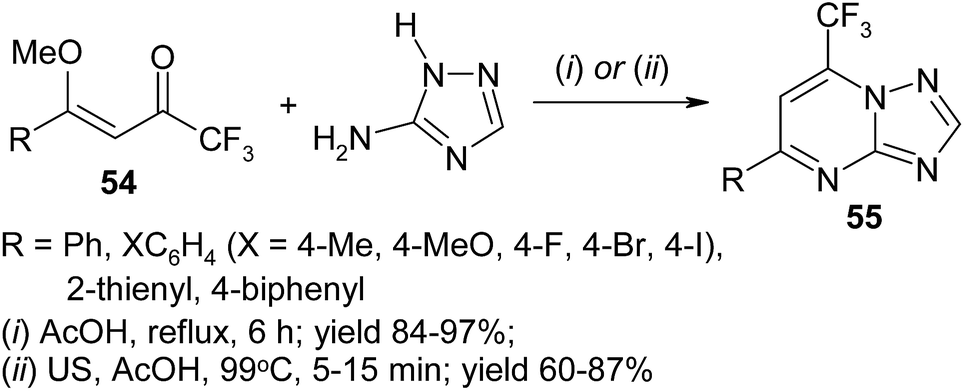 | ||
| Scheme 23 | ||
Finally, polyfluoroacylchromones 56 and their heteroaryl-analogues also can also be used as starting materials in the synthesis of functionally substituted polyfluoroalkylpyrimidines 57 (Scheme 24).107 Availability of initial reagents, experiment facility and good to high yields of target heterocycles indicate the appeal of this method. Interestingly, when the same chromones react with 1,3-C,N-binucleophiles (for example, ester, nitrile or amide of β-aminocrotonic acid), the novel polyfluoroalkylated nicotinic acid derivatives were obtained.108
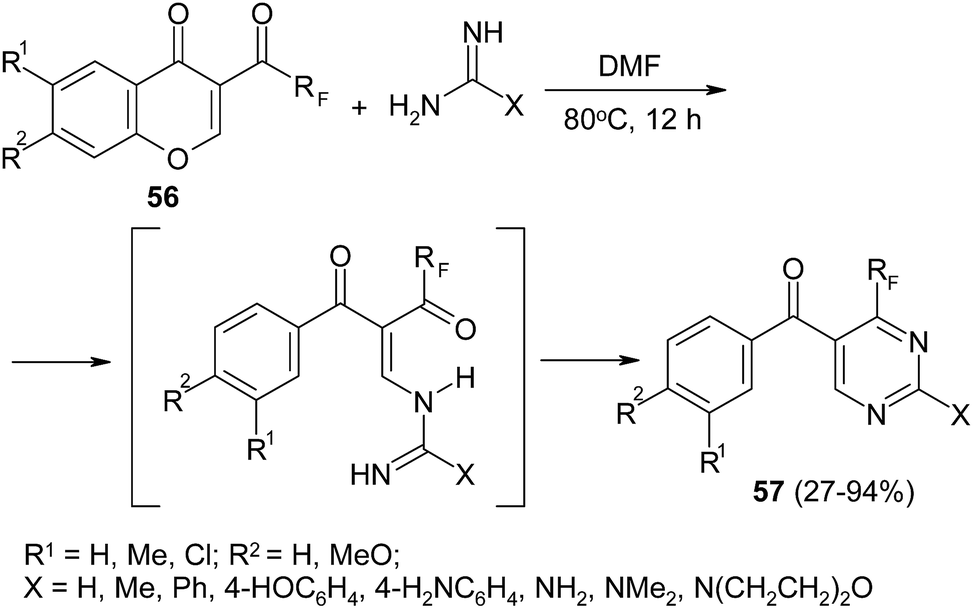 | ||
| Scheme 24 | ||
3.2. Quinolines
One of the classical approaches to the quinolines is based on the condensation reaction of substituted anilines with carbonyl compounds bearing active α-methylene component (Friedländer annulations). But its use in the synthesis of polyfunctional quinoline derivatives containing electron withdrawing polyhaloalkyl group was limited because the starting materials required for this method are rather difficult to obtain. This problem was solved when 2-aminoacetophenone 59 and polyhaloalkyated β-alkoxyenones 58 were used as initial reagents (Scheme 25). It was found that the best results were obtained in ionic liquid (IL) under microwave irradiation.109 In this case the target heterocycles were isolated in high yields (70–91%) in short time (10–20 minutes). Although equimolar amounts of ionic liquid and p-toluenesulfonic acid were used the quinolines 60 were isolated by chloroform extraction. After solvent evaporation no purification of reaction product was needed. | ||
| Scheme 25 | ||
The authors assume that reaction proceeds via formation of push–pull aminoenone which undergoes subsequent intramolecular cyclization and dehydration. At that the combination of ionic liquid and Brønsted acid have a catalytic effect on the final steps of all cascade of transformations.
3.3. Piperazines
Piperazine ring is known to have a special rank in medicinal chemistry because it is structural fragment of many biologically active compounds. The authors of papers,110,111 seeking for the shortest approach to trifluoroacetylated piperazines, have studied the reaction of CF3-bromoenones 61 with 1,2-diamines (Scheme 26). Ethylenediamine derivatives were supposed to react with enones 61a–e by classic aza-MIRC scheme including initial attack to β-olefinic carbon atom and subsequent nucleophilic substitution of halogen atom. However, the reaction of trifluoromethylated ketones 61a–e with symmetrically substituted ethylenediamine derivatives did not give the expected trifluoroacetylpiperazines 62. The authors, to their surprise, isolated isomeric piperazinones 63 bearing CF3-moiety at quaternary carbon atom of the cycle. This reaction proceeds smoothly, without any catalyst at room temperature. | ||
| Scheme 26 | ||
The NMR monitoring (1H, 19F, 13C) allowed the authors to register the formation of piperazinol 65 as key intermediate of all cascade of transformations and trace its transformation into final reaction product 63. These data formed the basis of the hypothesis of possible mechanism of rearrangement. The first step is the formation of captodative aminoalkene 64 by the classic scheme Ad–SN–E. The next step is intramolecular condensation with participation of the second amino group. The extra amino center of diamine directs the reaction towards piperazinol 65. In its absence (in the case of secondary monoamines) the reaction results in formation of indenole 66.112,113 The whole cascade of transformation finishes with formal 1,2-shift of trifluoromethyl group. According to proposed hypothesis, the polar solvents should promote the reaction and that was observed experimentally. Thus, it is trifluoroethanol that provides the highest yields of piperazinones 63 due to good balance of its polarity and acidity. The authors developed an approach to a very rare and hard to access type of piperazine derivatives and the rearrangement itself is the first example of easy migration of trifluoromethyl group to adjacent carbon atom.
Interestingly, among all N,N-binucleophiles there was one exception – dicyclopropyl derivative of ethylenediamine.111 In this case, instead of piperazinone 63 the isomeric trifluoroacetylpiperazine 62 was obtained (Scheme 27). Theoretical analysis showed that the main reason of different behavior of diamines consists in different solvation of transition states of nucleophilic substitution reaction of halogen atom.
 | ||
| Scheme 27 | ||
In contrast to symmetrically substituted ethylenediamines, their analogues containing two primary amino groups react with CF3-bromoenones 61a, b to give bicyclic derivatives 67 (Scheme 28).114 The reaction proceeds under mild conditions leading to 1,4-diazabicyclo[4.1.0]hept-4-enes 67 in good yield. Their synthesis is highly stereoselective: only one diastereoisomer is formed in all cases.
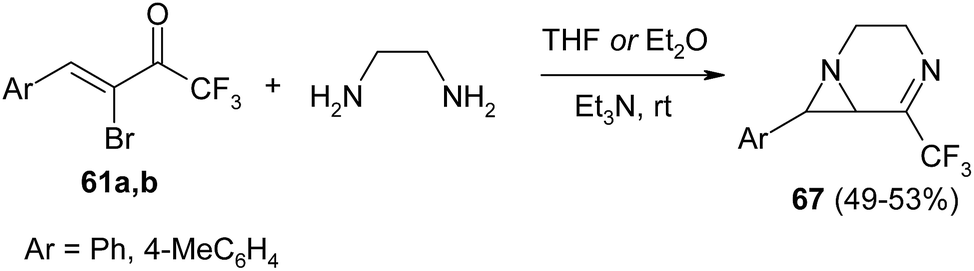 | ||
| Scheme 28 | ||
Less nucleophilic ortho-phenylenediamine (o-PDA) was also successfully involved in reaction with CF3-bromoenones 61a, b (Scheme 29). However, in this case the cascade of transformations stops at the stage of formation of hemiaminals 68 which were isolated in high yields.115
 | ||
| Scheme 29 | ||
The synthesis of heterocycles 67 and 68 confirms that reaction of bromoenones 61 with diamines bearing two amino groups proceeds by classic scheme involving such key steps as Michael addition of diamine, intramolecular substitution of bromine leading to formation of aziridine ring, and finally, closure of piperazine ring as a result of attack of second nucleophilic center on carbon atom of carbonyl group.116,117
4. Synthesis of seven-membered heterocycles
4.1. Diazepines
Non-fluorinated diazepines are generally obtained by condensation reaction of 1,2-diamines with acetylenic ketones118 or 1,3-diketones.119,120 However, CF3-diketones react in different way to give benzimidazoles, aminoenones, or macrocycles.121,122 The high polarity of triple bond as well as different nature of two electrophilic sp- and sp2-centers of polyfluoroalkylated acetylenic ketones favours selective synthesis of fluorine-containing diazepines which makes them more attractive substrates.Indeed, ethylenediamine reacts with CF3-ynones 5a, d leading to diazepines 69 in mild yield (Scheme 30). The formation of exclusively enamine tautomer seems to be due to the emergence of longer conjugation chain.123
 | ||
| Scheme 30 | ||
Similarly to ethylenediamine, o-phenylenediamine reacts with acetylenic CF3-ketones 5a, c, d to give benzodiazepines 70 in good yield (Scheme 31). The mild yield of hexyl-substituted diazepine (R = Hex) is explained by high content of push–pull aminoenone 71 in reaction mixture. According to NMR-spectroscopy, the latter is a key intermediate of all cascade of transformations.123
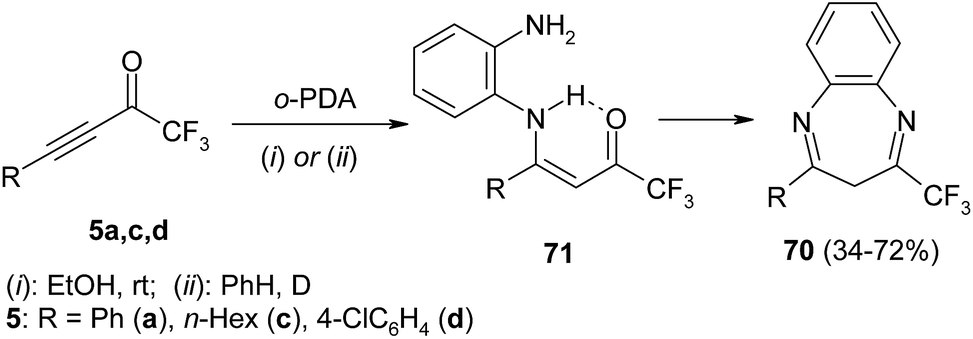 | ||
| Scheme 31 | ||
The proposed method of the synthesis of trifluoromethyl-containing [1,4]-diazepines is more attractive in comparison with that one based on 1,3-diketones: the target heterocycles are formed under mild conditions in good yield and with high selectivity.
Push–pull aminoenones 74 are formed on the first step of reaction of trifluoromethylated β-alkoxyenones 72 with 2,3-diaminopyridine 73 (Scheme 32). When reaction proceeds at low temperature (MeOH, 0 °C), they can be isolated in high yield.124 Subsequent long heating of reaction mixture in methanol leads to formation of diazepinols 75. The target heterocycles 75 can be obtained in one step from initial alkoxyenones in yield 54–71%.
 | ||
| Scheme 32 | ||
In contrast with CF3-enones, their trichloromethylated analogues react under similar conditions with 2,3-diaminopyridine to form corresponding diazepinones in good yield.125
It was reported earlier that CF3-enone 76 bearing two methoxy groups at β-position reacted with ortho-arylenediamines under microwave irradiation to give benzimidazoles instead of expected diazepines.126 One decade later Okada et al. repeated this reaction at room temperature127,128 and obtained diazepinols 77 with traces of corresponding diazepines 78 (Scheme 33). The efforts to carry out acid-catalyzed dehydratation of diazepinols 77 failed: in all cases the reaction led to formation of complex multi-component mixtures. The solution of this problem was found when diazepinols 77 were distilled in vacuo at the temperature 110–150 °C.127
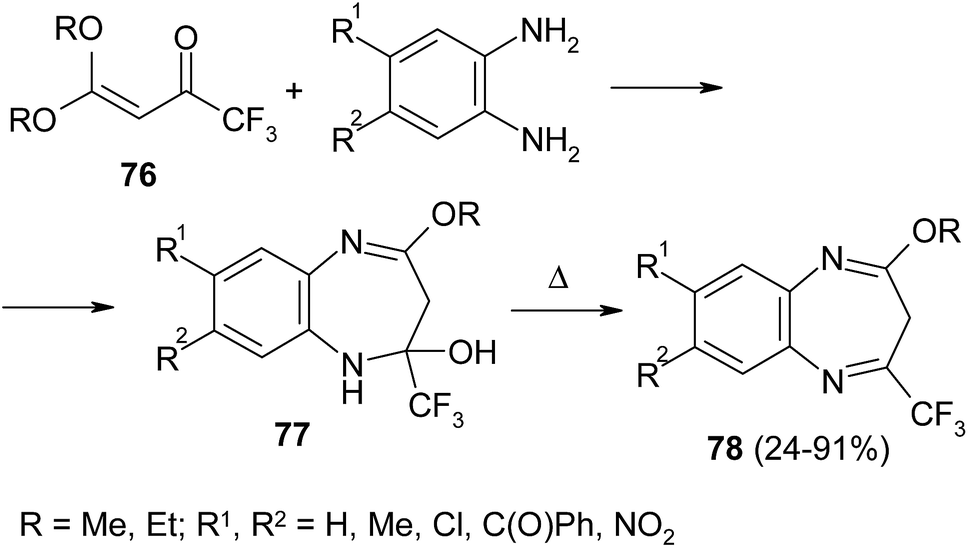 | ||
| Scheme 33 | ||
5. Conclusions and future outlook
Despite a number of procedures for the preparation of fluorinated nitrogen-bearing heterocycles, the development of new synthetic approaches to these compounds is still challenging research topic to the world chemical community. The future progress in the synthesis of such type of heterocyclic compounds will require the development of new starting materials. A significant number of new synthetic protocols based on the reactions of unsaturated polyfluoroalkylated ketones were elaborated during the last decade. Their understanding and wide application provides easy access to target heterocycles which sometimes cannot be prepared by other methods. There is no doubt that further research will enjoy much attention and unsaturated carbonyl compounds will play a major role in this field.Acknowledgements
This review has been written with the financial support of the Russian Foundation for Basic Research (Project No. 13-03-00063a).References
- V. Gouverneur and K. Seppelt, Chem. Rev., 2015, 115, 563 CrossRef CAS PubMed.
- M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- P. V. Ramachandran, Future Med. Chem., 2009, 1, 771 CrossRef CAS PubMed.
- R. Filler and R. Saha, Future Med. Chem., 2009, 1, 771 CrossRef PubMed.
- Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, John Wiley & Sons, Chichester, UK, 2009, pp. 640 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- W. L. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- J. P. Bégué and D. Bonnet-Delpon, in Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, Hoboken, 2008, pp. 72–98 Search PubMed.
- Fluorine and Health, Molecular Imaging, Biomedical Materials and Pharmaceuticals, ed. A. Tressaud and G. Haufe, Elsevier, Amsterdam, 2008, pp. 553–778 Search PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- A. Togni, Adv. Synth. Catal., 2010, 352, 2689 CrossRef CAS.
- Fluorine in Heterocyclic Chemistry, ed. V. G. Nenajdenko, Springer, 2014, vol. 1, pp. 681, V.2, 760 pp Search PubMed.
- Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications, ed. V. A. Petrov, Wiley, New Jersey, 2009, pp. 515 Search PubMed.
- Fluorinated Heterocycles, ed. A. A. Gakh and K. L. Kirk, ACS Symposium Series, Oxford University Press/American Chemical Society, Washington, DC, 2009, pp. 360 Search PubMed.
- V. Dinoiu, Rev. Roum. Chim., 2007, 52, 219 CAS.
- K. Uneyama, J. Fluorine Chem., 1999, 97, 11 CrossRef CAS.
- M. A. P. Martins, A. P. Sinhorin, A. Rosa, A. F. C. Flores, A. D. Wastowski, C. M. P. Pereira, D. C. Flores, P. Beck, R. A. Freitag, S. Brondani, W. Cunico, H. G. Bonacorso and N. Zanatta, Synthesis, 2002, 2353 CrossRef CAS.
- Domino Reactions: Concepts for Efficient Organic Synthesis, ed. L. F. Tietze, Wiley-VCH, 2014, pp. 648 Search PubMed.
- P. J. Parsons, C. S. Penkett and A. J. Shell, Chem. Rev., 1996, 96, 195 CrossRef CAS PubMed.
- S. V. Druzhinin, E. S. Balenkova and V. G. Nenajdenko, Tetrahedron, 2007, 63, 7753 CrossRef CAS.
- V. G. Nenajdenko, A. V. Sanin and E. S. Balenkova, Russ. Chem. Rev., 1999, 68, 483 CrossRef.
- V. G. Nenajdenko, A. V. Sanin and E. S. Balenkova, Molecules, 1997, 2, 186 CrossRef CAS.
- A. Y. Rulev, Russ. Chem. Rev., 2011, 80, 197 CrossRef CAS.
- I. S. Kondratov, V. G. Dolovanyuk, N. A. Tolmachova, I. I. Gerus, K. Bergander, R. Fröhlich and G. Haufe, Org. Biomol. Chem., 2012, 10, 8778 CAS.
- V. Kumar, R. Aggarwal and S. P. Singh, Heterocycles, 2008, 75, 2893 CrossRef.
- R. J. Linderman and K. S. Kirollos, Tetrahedron Lett., 1989, 30, 2049 CrossRef CAS.
- G. Ji, X. Wang, S. Zhang, Y. Xu, Y. Ye, M. Li, Y. Zhang and J. Wang, Chem. Commun., 2014, 50, 4361 RSC.
- M. A. P. Martins, D. J. Emmerich, C. M. P. Pereira, W. Cunico, M. Rossato, N. Zanatta and H. G. Bonacorso, Tetrahedron Lett., 2004, 45, 4935 CrossRef CAS.
- S. Li, Z. Li, D. Peng, Y. Li, J. Zhu, H. Xie, Y. Yuan, Z. Chen and Y. Wu, Chin. J. Chem., 2011, 29, 2695 CrossRef CAS.
- M.-T. Hsieh, S.-C. Kuo and H.-C. Lin, Adv. Synth. Catal., 2015, 357, 683 CrossRef CAS.
- Y. Wang, J. Han, J. Chen and W. Cao, Tetrahedron, 2015, 71, 8256 CrossRef CAS.
- D. N. Bazhin, D. L. Chizov, G.-V. Röschenthaler, Yu. S. Kudyakova, Ya. V. Bugart, P. A. Slepukhin, V. I. Saloutin and V. N. Charushin, Tetrahedron Lett., 2012, 55, 5714 CrossRef.
- M. A. P. Martins, A. P. Sinhorin, C. P. Frizzo, L. Buriol, E. Scapin, N. Zanatta and H. G. Bonacorso, J. Heterocycl. Chem., 2010, 50, 71 CrossRef.
- V. O. Iaroshenko, V. Specowius, K. Vlach, M. Vilches-Herrera, D. Ostrovskyi, S. Mkrtchyan, A. Villinger and P. Langer, Tetrahedron, 2011, 67, 5663 CrossRef CAS.
- D. L. Obydennov and B. I. Usachev, J. Fluorine Chem., 2012, 141, 41 CrossRef CAS.
- V. Ya Sosnovskikh, R. A. Irgashev, V. S. Moshkin and M. I. Kodess, Russ. Chem. Bull., Int. Ed., 2008, 57, 2146 CrossRef.
- V. M. Muzalevskiy, A. A. Iskandarov and V. G. Nenajdenko, Mendeleev Commun., 2014, 24, 342 CrossRef CAS.
- M. A. P. Martins, M. R. B. Marzari, C. P. Frizzo, M. Zanatta, L. Buriol, V. P. Andrade, N. Zanatta and H. G. Bonacorso, Eur. J. Org. Chem., 2012, 2012, 7112 CrossRef CAS.
- H. G. Bonacorso, C. A. Cechinel, J. Navarini, R. Andrighetto, M. A. P. Martins and N. Zanatta, Monatsh. Chem., 2013, 144, 1043 CrossRef.
- L. Buriol, C. P. Frizzo, L. D. T. Prola, D. N. Moreira, E. Scapin, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Catal. Lett., 2011, 141, 1130 CrossRef CAS.
- L. Buriol, C. P. Frizzo, M. R. B. Marzari, D. N. Moreira, L. D. T. Prola, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, J. Braz. Chem. Soc., 2010, 21, 1037 CrossRef CAS.
- T. N. Glasnov, K. Groschner and C. O. Kappe, ChemMedChem, 2009, 4, 1816 CrossRef CAS PubMed.
- D. Obermayer, T. N. Glasnov and C. O. Kappe, J. Org. Chem., 2011, 76, 6657 CrossRef CAS PubMed.
- M. A. P. Martins, C. M. P. Pereira, S. Moura, C. P. Frizzo, N. Zanatta, H. G. Bonacorso and A. F. C. Flores, J. Heterocycl. Chem., 2007, 44, 1195 CrossRef CAS.
- M. Rossatto, C. Casanova, A. P. Lima, D. J. Emmerich, V. Oliveira, R. M. Dallago, C. P. Frizzo, D. N. Moreira, L. Buriol, S. Brondani, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, ARKIVOC, 2014, ii, 224 Search PubMed.
- N. Zanatta, S. S. Amaral, J. M. dos Santos, A. M. P. W. da Silva, J. M. F. M. Schneider, L. da S. Fernandes, H. G. Bonacorso and M. A. P. Martins, Tetrahedron Lett., 2013, 54, 4076 CrossRef CAS.
- G. V. Bozhenkov, V. A. Savosik, L. I. Larina, L. V. Klyba, E. R. Zhanchipova, A. N. Mirskova and G. G. Levkovskaya, Russ. J. Org. Chem., 2008, 44, 1014 CrossRef CAS.
- M. A. Barabanov, D. V. Sevenard and V. Y. Sosnovskikh, Russ. Chem. Bull., Int. Ed., 2012, 61, 1646 CrossRef CAS.
- R. T. Iminov, A. V. Mashkov, I. I. Vyzir, B. A. Chalyk, A. V. Tverdokhlebov, P. K. Mykhailiuk, L. N. Babichenko, A. A. Tolmachev, Y. M. Volovenko, A. Biitseva, O. V. Shishkin and S. V. Shishkina, Eur. J. Org. Chem., 2015, 2015, 886 CrossRef CAS.
- J. S. Scott, J. de Schoolmeester, E. Kilgour, R. M. Mayers, M. J. Packer, D. Hargreaves, S. Gerhardt, D. J. Ogg, A. Rees, N. Selmi, A. Stocker, J. G. Swales and P. R. O. Whittamore, Eur. J. Med. Chem., 2012, 55, 10136 CrossRef CAS PubMed.
- M. V. Pryadeina, A. B. Denisova, Y. V. Burgart and V. I. Saloutin, Russ. J. Org. Chem., 2008, 44, 1811 CrossRef CAS.
- H. G. Bonacorso, L. M. F. Porte, C. A. Cechinel, G. R. Paim, E. D. Deon, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2009, 50, 1392 CrossRef CAS.
- H. G. Bonacorso, C. W. Wiethan, L. M. F. Porte, M. C. Moraes, J. Navarini, C. R. Belo, F. M. Luz, N. Zanatta and M. A. P. Martins, ARKIVOC, 2013, iv, 291 Search PubMed.
- A. F. C. Flores, L. A. Piovesan, L. Pizzuti, D. C. Flores, J. L. Malavolta and M. A. P. Martins, J. Heterocycl. Chem., 2014, 51, 733 CrossRef CAS.
- H. G. Bonacorso, R. P. Vezzosi, I. R. Rodrigues, R. L. Drekener, L. M. F. Porte, A. F. C. Flores, N. Zanatta and M. A. P. Martins, J. Braz. Chem. Soc., 2009, 20, 1370 CrossRef CAS.
- H. G. Bonacorso, L. M. F. Porte, G. R. Paim, F. M. Luz, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2010, 51, 3759 CrossRef CAS.
- H. G. Bonacorso, C. A. Cechinel, L. M. F. Porte, J. Navarini, S. Cavinatto, R. C. Sehnem, D. B. Martins, N. Zanatta and M. A. P. Martins, J. Heterocycl. Chem., 2010, 47, 1073 CrossRef CAS.
- H. G. Bonacorso, S. Cavinatto, P. T. Campos, L. M. F. Porte, J. Navarini, G. R. Paim, M. A. P. Martins, N. Zanatta and C. Z. Stuker, J. Fluorine Chem., 2012, 125, 303 CrossRef.
- H. G. Bonacorso, C. A. Cechinel, E. D. Deon, R. C. Sehnem, F. M. Luz, M. A. P. Martins and N. Zanatta, ARKIVOC, 2009, ii, 174 Search PubMed.
- A. F. C. Flores, P. F. Rosales, J. l. Malavolta and D. C. Flores, J. Braz. Chem. Soc., 2014, 25, 1439 CAS.
- P. E. Almeida da Silva, D. F. Ramos, H. G. Bonacorso, A. I. de la Iglesia, M. R. Oliveira, T. Coelho, J. Navarini, H. R. Morbidoni, N. Zanatta and M. A. P. Martins, Int. J. Antimicrob. Agents, 2008, 32, 139 CrossRef CAS.
- H. G. Bonacorso, E. P. Pittaluga, S. H. Alves, L. F. Schaffer, S. Cavinatto, L. M. F. Porte, G. R. Paim, M. A. P. Martins and N. Zanatta, ARKIVOC, 2012, viii, 62 CAS.
- P. Machado, F. A. Rosa, M. Rossatto, G. da S. Sant'Anna, P. D. Sauzem, R. M. Siqueira da Silva, M. A. Rubin, J. Ferreira, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, ARKIVOC, 2007, xvi, 281 Search PubMed.
- D. N. Moreira, C. P. Frizzo, K. Longhi, A. B. Soares, M. R. B. Marzari, L. Buriol, S. Brondani, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Monatsh. Chem., 2011, 142, 1265 CrossRef CAS.
- H. G. Bonacorso, G. R. Paim, C. Z. Guerra, R. C. Sehnem, C. A. Cechinel, L. M. F. Porte, M. A. P. Martins and N. Zanatta, J. Braz. Chem. Soc., 2009, 20, 509 CrossRef CAS.
- H. G. Bonacorso, C. A. Cechinel, J. Navarini, R. Andrighetto, M. A. P. Martins and N. Zanatta, Monatsh. Chem., 2011, 142, 277 CrossRef CAS.
- H. G. Bonacorso, C. A. Cechinel, E. P. Pittaluga, A. Ferla, L. M. F. Porte, M. A. P. Martins and N. Zanatta, J. Braz. Chem. Soc., 2010, 21, 1656 CrossRef CAS.
- D. N. Moreira, C. P. Frizzo, K. Longhi, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Monatsh. Chem., 2008, 139, 1049 CrossRef CAS.
- P. D. Sauzem, P. Machado, M. A. Rubin, G. da S. Sant'Anna, H. B. Faber, A. H. de Souza, C. F. Mello, P. Beck, R. A. Burrow, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Eur. J. Med. Chem., 2008, 43, 1237 CrossRef CAS PubMed.
- M. A. P. Martins, P. H. Beck, D. N. Moreira, L. Buriol, C. P. Frizzo, N. Zanatta and H. G. Bonacorso, J. Heterocycl. Chem., 2010, 47, 301 CAS.
- D. N. Moreira, C. P. Frizzo, K. Longhi, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Monatsh. Chem., 2011, 142, 515 CrossRef.
- M. A. P. Martins, D. N. Moreira, C. P. Frizzo, K. Longhi, N. Zanatta and H. G. Bonacorso, J. Braz. Chem. Soc., 2008, 19, 1361 CrossRef CAS.
- H. G. Bonacorso, M. S. Correa, L. M. F. Porte, E. P. Pittaluga, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2012, 53, 5488 CrossRef CAS.
- M. A. P. Martins, P. Machado, F. A. Rosa, W. Cunico, H. G. Bonacorso and N. Zanatta, Mini-Rev. Org. Chem., 2008, 5, 53 CrossRef CAS.
- K. V. Tarasenko, O. V. Manoylenko, V. P. Kukhar, G.-V. Röschenthaler and I. I. Gerus, Tetrahedron Lett., 2010, 51, 4623 CrossRef CAS.
- M. A. P. Martins, G. M. Siqueira, G. P. Bastos, H. G. Bonacorso and N. Zanatta, J. Heterocycl. Chem., 1996, 33, 1619 CrossRef CAS.
- M. A. P. Martins, P. Machado, L. A. Piovesan, A. F. C. Flores, M. M. A. de Campos, C. Scheidt, H. G. Bonacorso and N. Zanatta, Monatsh. Chem., 2008, 139, 985 CrossRef CAS.
- V. Y. Sosnovskikh, V. S. Moshkin and M. I. Kodess, Tetrahedron, 2008, 64, 7877 CrossRef CAS.
- H. Jiang, W. Yue, H. Xiao and S. Zhu, Tetrahedron, 2007, 63, 2315 CrossRef CAS.
- H. G. Bonacorso, M. B. Costa, C. A. Cechinel, R. C. Sehnem, M. A. P. Martins and N. Zanatta, J. Heterocycl. Chem., 2009, 46, 158 CrossRef CAS.
- C. P. Frizzo, M. R. B. Marzari, C. R. Bender, I. M. Grindi, J. Trindade, L. Buriol, G. S. Caleffi, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Monatsh. Chem., 2014, 145, 797 CrossRef CAS.
- N. Zanatta, C. C. Madruga, P. C. Marisco, L. S. da Rosa, F. M. da Silva, H. G. Bonacorso and M. A. P. Martins, J. Heterocycl. Chem., 2010, 47, 1234 CrossRef CAS.
- N. Zanatta, C. C. Madruga, P. C. Marisco, L. S. da Rosa, L. da S. Fernandes, D. C. Flores, A. F. C. Flores, R. A. Burrow, H. G. Bonacorso and M. A. P. Martins, J. Heterocycl. Chem., 2008, 45, 221 CrossRef CAS.
- H. G. Bonacorso, A. Ferla, C. A. Cechinel, N. Zanatta and M. A. P. Martins, J. Heterocycl. Chem., 2008, 45, 483 CrossRef CAS.
- N. Zanatta, S. S. Amaral, J. M. dos Santos, D. L. de Mello, L. da S. Fernandes, H. G. Bonacorso, M. A. P. Martins, A. D. Andricopulo and D. M. Borchhardt, Bioorg. Med. Chem., 2008, 16, 10236 CrossRef CAS PubMed.
- H. G. Bonacorso, G. P. Bortolotto, J. Navarini, L. M. F. Porte, C. W. Wiethan, N. Zanatta, M. A. P. Martins and A. F. C. Flores, J. Fluorine Chem., 2010, 131, 1297 CrossRef CAS.
- V. Gressler, S. Moura, A. F. C. Flores, D. C. Flores, P. Colepicolo and E. Pinto, J. Braz. Chem. Soc., 2010, 21, 1477 CrossRef CAS.
- M. V. Goryaeva, Y. V. Burgart and V. I. Saloutin, Russ. Chem. Bull., 2009, 58, 1259 CrossRef CAS.
- N. Zanatta, D. Faoro, L. da S. Fernandes, P. B. Brondani, D. C. Flores, A. F. C. Flores, H. G. Bonacorso and M. A. P. Martins, Eur. J. Org. Chem., 2008, 2008, 5832 CrossRef.
- A. F. C. Flores, L. Pizzuti, S. Brondani, M. Rossato, N. Zanatta and M. A. P. Martins, J. Braz. Chem. Soc., 2007, 18, 1316 CrossRef CAS.
- A. F. C. Flores, J. L. Malavolta, A. A. Souto, R. B. Goularte, D. C. Flores and L. A. Piovesan, J. Braz. Chem. Soc., 2013, 24, 580 CAS.
- D. R. Fandrick, D. Reinhardt, J.-N. Desrosiers, S. Sanyal, K. R. Fandrick, S. Ma, N. Grinberg, H. Lee, J. J. Song and C. H. Senanayake, Org. Lett., 2014, 16, 2834 CrossRef CAS PubMed.
- F. A. Rosa, P. Machado, G. F. Fiss, P. S. Vargas, T. S. Fernandes, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Synthesis, 2008, 3639 CAS.
- V. O. Iaroshenko, S. Dudkin, V. Y. Sosnovskikh, A. Villinger and P. Langer, Eur. J. Org. Chem., 2013, 2013, 3166 CrossRef CAS.
- A. F. C. Flores, D. C. Flores, G. Oliveira, L. Pizzuti, R. M. S. da Silva, M. A. P. Martins and H. G. Bonacorso, J. Braz. Chem. Soc., 2008, 19, 184 CrossRef CAS.
- M. V. Goryaeva, Y. V. Burgart and V. I. Saloutin, J. Fluorine Chem., 2013, 147, 15 CrossRef CAS.
- L. Buriol, T. S. München, C. P. Frizzo, M. R. B. Marzari, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Ultrason. Sonochem., 2013, 20, 1139 CrossRef CAS PubMed.
- E. E. Emelina and A. A. Petrov, Russ. J. Org. Chem., 2009, 45, 417 CrossRef CAS.
- M. V. Pryadeina, Y. V. Burgart, V. I. Saloutin, P. A. Slepukhin, E. V. Sadchikova and E. N. Ulomskii, Russ. J. Org. Chem., 2009, 45, 242 CrossRef CAS.
- C. P. Frizzo, E. Scapin, M. R. B. Marzari, T. S. München, N. Zanatta, H. G. Bonacorso, L. Buriol and M. A. P. Martins, Ultrason. Sonochem., 2014, 21, 958 CrossRef CAS PubMed.
- M. V. Goryaeva, Y. V. Burgart, V. I. Saloutin, E. V. Sadchikova and E. N. Ulomskii, Heterocycles, 2009, 78, 435 CrossRef CAS.
- M. V. Pryadeina, Y. V. Burgart, V. I. Saloutin and O. N. Chupakhin, Mendeleev Commun., 2008, 18, 276 CrossRef CAS.
- M. V. Goryaeva, Y. V. Burgart and V. I. Saloutin, Russ. J. Org. Chem., 2010, 46, 432 CrossRef CAS.
- M. V. Goryaeva, Y. V. Burgart, V. I. Saloutin and O. N. Chupakhin, Heterocycl. Compd., 2012, 48, 372 CrossRef CAS.
- A. Kotljarov, R. A. Irgashev, V. O. Iaroshenko, D. V. Sevenard and V. Y. Sosnovskikh, Synthesis, 2009, 3233 CAS.
- V. Y. Sosnovskikh, R. A. Irgashev and M. I. Kodess, Tetrahedron, 2008, 64, 2997 CrossRef CAS.
- L. D. T. Prola, L. Buriol, C. P. Frizzo, G. S. Caleffi, M. R. B. Marzari, D. N. Moreira, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, J. Braz. Chem. Soc., 2012, 23, 1663 CrossRef CAS.
- A. Y. Rulev, V. M. Muzalevskiy, E. V. Kondrashov, I. A. Ushakov, A. R. Romanov, V. N. Khrustalev and V. G. Nenajdenko, Org. Lett., 2013, 15, 2726 CrossRef CAS PubMed.
- V. M. Muzalevskiy, Y. A. Ustynyuk, I. P. Gloriosov, V. A. Chertkov, A. Y. Rulev, E. V. Kondrashov, I. A. Ushakov, A. R. Romanov and V. G. Nenajdenko, Chem.–Eur. J., 2015, 21, 16982 CrossRef CAS PubMed.
- A. Y. Rulev, I. A. Ushakov, V. G. Nenajdenko, E. S. Balenkova and M. G. Voronkov, Eur. J. Org. Chem., 2007, 6039 CrossRef CAS.
- A. Y. Rulev, I. A. Ushakov and V. G. Nenajdenko, Tetrahedron, 2008, 64, 8073 CrossRef CAS.
- (a) V. M. Muzalevskiy, A. Y. Rulev, E. V. Kondrashov, A. R. Romanov, I. A. Ushakov, V. A. Chertkov and V. G. Nenajdenko, Eur. J. Org. Chem, 2016 Search PubMed submitted; (b) A. Y. Rulev and A. R. Romanov, Aza-MIRC Reaction: An Efficient Cascade Assembly of N-Heterocycles, Abstracts of 3rd International Congress on Heterocyclic Chemistry, Moscow, 2015, p. 121 Search PubMed.
- A. V. Fokin, A. F. Kolomiets and N. V. Vasil'ev, Russ. Chem. Rev., 1984, 53, 238 CrossRef.
- A. Y. Rulev and J. Maddaluno, Phys. Org. Chem., 2002, 15, 590 CrossRef CAS.
- A. Y. Rulev, Russ. Chem. Rev., 1998, 67, 279 CrossRef.
- S.-G. Huang, H.-F. Mao, S.-F. Zhou, J.-P. Zou and W. Zhang, Tetrahedron Lett., 2013, 54, 6178 CrossRef CAS.
- A. Gharib, M. Jahangir and J. W. Scheeren, Synth. Commun., 2013, 43, 309 CrossRef CAS.
- B. R. Vaddula, R. S. Varma and J. Leazer, Tetrahedron Lett., 2013, 54, 1538 CrossRef CAS.
- M. Narsaiah, R. Rao, R. Reddy, S. Rao and V. R. Yadla, J. Fluorine Chem., 2003, 124, 203 CrossRef.
- V. Charushin, D. Chizhov, V. Filyakova, E. Khmara, M. Pervova, V. Saloutin and M. Samorukova, J. Fluorine Chem., 2011, 132, 394 CrossRef.
- A. R. Romanov, A. Y. Rulev, I. A. Ushakov, V. M. Muzalevskiy and V. G. Nenajdenko, Mendeleev Commun., 2014, 24, 269 CrossRef CAS.
- H. G. Bonacorso, R. V. Lourega, F. J. Righi, E. D. Deon, N. Zanatta and M. A. P. Martins, J. Heterocycl. Chem., 2008, 45, 1679 CrossRef CAS.
- H. G. Bonacorso, R. V. Lourega, E. D. Deon, N. Zanatta and M. A. P. Martins, Tetrahedron Lett., 2007, 48, 4835 CrossRef CAS.
- A. C. S. Reddy, P. S. Rao and R. V. Venkataratnam, Tetrahedron, 1997, 53, 5847 CrossRef CAS.
- N. Ota, E. Okada, N. Terai, T. Miyamura, D. Shibata and T. Sakata, Heterocycles, 2009, 77, 983 CrossRef CAS.
- N. Ota, T. Tomoda, N. Terai, Y. Kamitori, D. Shibata, M. Médebielle and E. Okada, Heterocycles, 2008, 76, 1205 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |