
Preparation of cellulose-graft-polylactic acid via melt copolycondensation for use in polylactic acid based composites: synthesis, characterization and properties
Sun Hua,
Feng Chen,
Zheng-ying Liu,
Wei Yang and
Ming-bo Yang*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, Sichuan, People's Republic of China. E-mail: yangmb@scu.edu.cn
First published on 21st December 2015
Abstract
In order to improve the melt strength of poly(lactic acid) (PLA), microcrystalline cellulose-graft-polylactic acid (MCC-g-PLA) copolymer was prepared and introduced into the PLA matrix. The MCC-g-PLA copolymers were synthesized by melt copolycondensation of lactic acid (LA) with microcrystalline cellulose (MCC) which was pretreated to improve accessibility. The MCC-g-PLA copolymers with a molar substitution (MS) of PLA in the range of 1.67–5.97 were synthesized by adjusting reaction temperature, molar ratio of LA monomer to MCC and washing times in the pretreatment process. Compared with MCC, the crystalline structures of MCC-g-PLA copolymers were not perfect due to the existence of PLA side chains. A glass transition temperature (Tg) appeared in the copolymers, which had never been observed in unmodified MCC, and decreased with the increase of MS. When MS was above 4.41, the thermal degradation of PLA side chains emerged. When MCC-g-PLA copolymer was introduced into the PLA matrix, good dispersion of MCC-g-PLA was verified with SEM results. The extension rheology results showed that the melt strength of PLA can be effectively enhanced with the addition of MCC-g-PLA, especially at low elongation rate. Meanwhile, MCC-g-PLA also improved the crystallization ability of PLA in the non-isothermal crystallization process.
1. Introduction
Poly(lactic acid) (PLA), which holds the potential to replace traditional petroleum based polymer materials because of its excellent mechanical properties, outstanding processing performance, availability from renewable agricultural products and biodegradability, has gained enormous attention.1–3 It has been widely used in academic research and industrial applications,4–7 for example stretch-blown bottles,8 scaffold materials9–11 and so on. In addition to these applications, it is noteworthy that PLA could be used for plastic bags and plastic film due to its biodegradability, taking the environment problems into consideration.12 Taking agricultural plastic film as an example, the residue of agricultural plastic film in the field has already become an important negative factor that affects the agricultural environment, which destroys the soil structure and harms growth of crops, due to it being non-degradable.13 So it is of great value to use PLA film instead of traditional plastic film for environmental and demanding markets.12Although PLA possesses many advantages, the drawbacks of the poor viscoelastic behavior, low melt strength, the inherent brittleness, and slow crystallization rate, have limited its large-scale commercialization especially as film materials.14–16 As is known, blow molding, by which a bi-axial stress is imposed on the film bubble, is the most convenient and high-performance-price-ratio method to manufacture films. From such a process, impact properties of the film are excellent and mechanical and optical properties can be controlled.17 However, polymer melt undergoes a complex three-dimensional deformation process during blow molding, so to prepare blown film steadily and successfully, high melt strength is the basic requirement for materials, such as the case for branched low density polyethylene (LDPE). Unfortunately, the blow molding of PLA is quite difficult owing to its poor viscoelastic behavior and low melt strength. The brittleness of PLA also prevents it from being used as plastic bags and plastic film. Therefore, in order to improve the processability of PLA to be used as blown films and to prepare PLA films with good properties, PLA needs to be modified to improve the melt strength, viscoelastic behavior and toughness.16,18
Increasing the molecular weight and introducing long branched chains have been proved to be effective methods to improve the melt strength of PLA, which can increase chain entanglement and extend melt relaxation time of PLA chains. Although high molecular weight (Mw) PLA can be synthesized by ring-opening polymerization of lactide and chain extension in laboratory studies,19,20 Mw of commercial PLA is still not high enough to ensure the melt strength needed for blow molding. Introducing the long branched chains is another effective method, which also has a great influence on the viscoelastic behavior of polymer melt, and has been proved to be able to increase the melt strength and chain entanglement.21–23 So, if we can synthesized copolymer with long branched chains of PLA, the existence of PLA branched chains can not only increase entanglement of PLA chains and melt strength of PLA, but also improve the dispersion state of the copolymer in PLA, thus making PLA easy to process.
Cellulose is the most abundant, biodegradable, and renewable natural polymer in the earth, and holds many attractive properties, such as low cost, nontoxic, good mechanical properties and low density.24,25 If we can graft long branched chains of PLA onto cellulose, the biodegradable property of products is guaranteed, simultaneously, the applications of PLA and cellulose are both widened.
Graft copolymerization is a common way to introduce other polymers onto cellulose or cellulose derivatives.26–32 Unfortunately, cellulose has strong intramolecular and intermolecular hydrogen-bonding network and high crystallinity, which make it non-thermoplastic and insoluble in common solvent. Most reaction reagents penetrate only into the amorphous regions of cellulose,33 while a lot of hydroxyl groups are sealed in crystalline region, then it is difficult to access to reagents and participate in reaction. These restrict the reaction activity of cellulose. In recent years, new solvents of cellulose have been discovered, such as lithium chloride/N,N-dimethylacetamide and ionic liquid.34–36 They had been used as reaction media to graft polymer chains onto cellulose,37–42 which could improve the accessibility and reactivity of cellulose. But the use of solvents could bring some problems, such as toxicity, complicated preparation process, or degradability.
How can we improve the accessibility of cellulose in non-toxic solvent? According to previous studies,43–46 treating cellulose with aqueous sodium hydroxide (NaOH) solutions had a considerable effect on the hydrogen-bonding network and crystalline structure of cellulose, in this case the crystalline structure of cellulose can transform from cellulose I to cellulose II. In this process, the alkali can destroy the hydrogen bonds and infiltrate into the crystalline region, which could be utilized to improve the accessibility and activity of cellulose. Then the solvent can be exchanged. Thus, the reaction monomer could get inside of cellulose.
Because lactide is easy to decompose, lactic acid was chosen as reaction monomer. In this work, firstly we improved the accessibility of cellulose by solvent exchanged method. Then, melt copolycondensation of lactic acid with microcrystalline cellulose (MCC) was carried out to obtain microcrystalline cellulose-graft-polylactic acid copolymers using tin(II) chloride dehydrate catalyst. Finally, the synthesized copolymer was added into PLA matrix, and the properties of PLA composites were studied.
2. Experimental
2.1. Materials
Microcrystalline cellulose (MCC) with a degree of polymerization (DP) of ca. 200 was purchased from ChengDu KeLong Chemical Co., Ltd. China. Lactic acid (chemical grade, solute concentration: 85 wt%), sodium hydroxide (NaOH), tin(II) chloride dihydrate (SnCl2), toluene, dichloromethane, dimethyl sulfoxide (DMSO) and alcohol were reagent grade and purchased from ChengDu HaiHong Chemical Co., Ltd. China. PLA (trade name REVODE110) with a number-average molecular weight of 4 × 104 g mol−1 was purchased from Zhejiang Hisun Biomaterials Co., Ltd. China.2.2. Introduction of lactic acid to the inside of microcrystalline cellulose
A dissolving procedure of MCC in alkaline solution was performed as follows.47 2.5 g NaOH was dissolved in 26.9 g water at room temperature. 1 g MCC was then added into the mixture, resulting in a suspension of MCC in an 8.5 wt% NaOH solution. The suspension was put into refrigerator until it became a frozen solid state. The frozen solid was then put into room temperature to thaw, and 20.6 g water was added to the thawed mixture with gentle stirring, and a clean solution was obtained. The dissolved cellulose was precipitated with lactic acid and filtrated, and then the precipitate was washed with LA for 4 times, dried in an oven at 60 °C, and was coded as MCC-L. The weight of MCC-L was 26.2 g.As a contrast, using the same process, we used water to precipitate the dissolved cellulose and wash the precipitate, then the product was dried in an oven at 60 °C, and was coded as MCC-W. The weight of MCC-W was 0.9 g.
2.3. Synthesis of MCC-g-PLA copolymers
The undried MCC-L and lactic acid monomer (molar ratio of LA/AHG was 36/1) were put into a three-neck flask equipped with a mechanical stirrer and a thermometer. The system was heated in an oil bath and dehydrated under vacuum condition at 100 °C, 110 °C and 120 °C for 2 h, respectively. After dehydration, the temperature was heated up to initial reaction temperature, and then catalyst SnCl2 was added into flask. Then the absolute pressure was reduced to 500–700 Pa, and the melt copolycondensation was carried out by heating the reaction system at a gradient style to prevent the evaporation of LA molecules in early stage (specific reaction temperature and time was in the order as follows: 130 °C, 2 h; 140 °C, 2 h; 150 °C, 2 h; 160 °C, 2 h). After the reaction finished, 200 ml dichloromethane was poured into the flask. The resultant polymer was precipitated with alcohol, and then was extracted with toluene in a Soxhlet extraction apparatus for 24 h to remove the byproduct (homo-PLLA) formed in reaction. The purified product was soaked with dichloromethane for hours, and was filtered until the filtrate became transparent and no precipitate appeared when a few drops of filtrate were put into alcohol, which could confirm that no byproduct remained and the purification was sufficient. Finally the product, MCC-g-PLA, was dried in an oven at 60 °C for over 24 h, and was coded as MP-C2W4-T160, in which C represented the content of LA, W represented the washing times with LA in pretreatment, and T represented the maximum reaction temperature. Using the same process and adjusting reaction conditions or molar ratios of LA monomer to anhydroglucose (AHG), a series of MCC-g-PLA copolymers were prepared. The reaction conditions, molar ratios of LA monomer to MCC and reaction results were summarized in Table 1.| Sample | Washing times | LAa/AHG (mol mol−1) | T (°C) | DPs | DS | MS |
|---|---|---|---|---|---|---|
| a Indicates the amount added in melt copolycondensation process, which does not include the LA remained in MCC-L.b Specific reaction temperature and time: 120 °C, 2 h; 130 °C, 6 h.c Specific reaction temperature and time: 120 °C, 2 h; 130 °C, 2 h; 140 °C, 4 h.d Specific reaction temperature and time: 130 °C, 2 h; 140 °C, 2 h; 150 °C, 4 h. | ||||||
| MP-C0W4-T130 | 4 | 0 | 130b | 2.79 | 0.60 | 1.67 |
| MP-C0W4-T140 | 4 | 0 | 140c | 3.40 | 0.60 | 2.04 |
| MP-C0W4-T150 | 4 | 0 | 150d | 3.47 | 0.64 | 2.22 |
| MP-C0W4-T160 | 4 | 0 | 160 | 4.67 | 0.67 | 3.13 |
| MP-C1W4-T160 | 4 | 18/1 | 160 | 5.17 | 0.71 | 3.67 |
| MP-C2W4-T160 | 4 | 36/1 | 160 | 4.60 | 0.64 | 2.94 |
| MP-C2W3-T160 | 3 | 36/1 | 160 | 4.70 | 0.88 | 4.14 |
| MP-C2W2-T160 | 2 | 36/1 | 160 | 5.90 | 0.88 | 5.19 |
| MP-C2W1-T160 | 1 | 36/1 | 160 | 6.22 | 0.96 | 5.97 |
2.4. Preparation of PLA/MCC-g-PLA composites
Composites of PLA with MCC-g-PLA were prepared by solution mixing. PLA and MCC-g-PLA (sample MP-C2W1-T160 was used) were dissolved separately in DMSO before mixing. The concentrations of PLA/DMSO and MCC-g-PLA/DMSO were 3 g/100 ml and 3 g/1000 ml respectively. The two solutions were then mixed and the mixture was ultrasonically treated for 30 min. Subsequently, the mixture was precipitated in alcohol and dried to a constant weight at 60 °C in a vacuum oven, and finally the composites PLA/MCC-g-PLA were obtained. The composites were coded as PLA-MP-X, in which X represented the weight of MCC-g-PLA in 100 g PLA matrix and X was 2, 3, and 4, respectively. As a contrast, composites of PLA with unmodified MCC were prepared using the same process, and were coded as PLA-MCC-X, in which X represented the weight of MCC in 100 g PLA matrix, and X was 2, 3, and 4, respectively.2.5. Characterization
The thermal analysis of PLA/MCC-g-PLA composites was also performed by DSC. The samples of 5–7 mg were dried at 60 °C for 24 h before measurement, and were heated from 25 °C to 200 °C at a heating rate of 10 °C min−1 and kept at 200 °C for 3 min to eliminate the thermal history, then cooled to 25 °C at a rate of 5 °C min−1, and finally were reheated to 200 °C at a rate of 10 °C min−1 under nitrogen atmosphere. The cooling and second heating curves were recorded.
3. Results and discussion
3.1. Synthesis and characterization of MCC-g-PLA copolymers
WAXD spectra of MCC, MCC-W and MCC-L were shown in Fig. 1. Cellulose I and cellulose II are the most commonly used cellulose, and in WAXD spectra cellulose I shows three characteristic diffraction peaks at around 2θ = 14.8, 16.3 and 22.6°, whereas cellulose II shows three peaks at around 2θ = 12.1, 19.8 and 22.0°.43 From Fig. 1, MCC was a typical cellulose I, and MCC-W was a typical cellulose II. But MCC-L was neither cellulose I nor cellulose II, only a dispersive broad peak at 2θ = 20.0° was observed. | ||
| Fig. 1 WAXD spectra of MCC, MCC-W and MCC-L. | ||
In cellulose, the hydrogen-bonding network makes molecular chains arrange regularly and form crystal. Different from cellulose I with parallel chain structure, cellulose II was comprised of an array of antiparallel chain molecules, and the unit cell was monoclinic according to Kolpak and Blackwell.44 When MCC was treated in NaOH solution, alkali reached to amorphous and crystalline regions of MCC, and destroyed the hydrogen bonds in the crystalline regions, then MCC was dissolved in NaOH solution.47 When dissolved MCC was regenerated in water, the conformation of hydroxymethyl group changed,43–45 and cellulose I transformed to cellulose II with an antiparallel chain structure. So MCC-W was cellulose II. But for cellulose II, the hydrogen-bonding network and structure were also very dense, and the reaction reagents were still difficult to penetrate into the inside of cellulose and react with cellulose. So, simple alkali treatment could not improve the accessibility of cellulose. Therefore, if we wanted to introduce reaction reagents into cellulose, we had to use the stage when hydrogen bonds were destroyed.
According to the above analysis, after MCC was dissolved in NaOH solution, hydrogen bonds in cellulose were destroyed by alkali. At this time, if the reaction reagents were added into the mixture, reaction reagents may enter into the cellulose before the hydrogen bonds regenerated. For MCC-L, perfect crystalline structure was not observed, indicating that the crystal structure and hydrogen-bonding network had been disrupted. This may be owing to that adding lactic acid into the dissolved MCC solution did make lactic acid enter into cellulose, and the lactic acid remained in cellulose after drying at 60 °C. The boiling point of lactic acid was about 122 °C,48 and it was not easy to evaporate at 60 °C. This was also confirmed by increase in the weight of MCC-L compared with MCC-W. The remained LA molecules formed steric hindrance to hinder the configuration change of C–O bond at C6 position and the formation of hydrogen bonds, and broke the regularity of hydrogen-bonding network in cellulose, thus MCC-L cannot crystallize. That is, lactic acid had reached into the crystalline region of cellulose, and had a possibility of reacting with hydroxyl groups both in amorphous and crystalline region, and the accessibility of cellulose to lactic acid was improved.
The structure of MCC-g-PLA copolymers was characterized by FTIR and the results were presented in Fig. 2. Because the byproduct PLA homopolymer produced in melt copolycondensation process had been removed by purification process, the FTIR spectra should reflect the chemical structure of the grafted copolymers. As shown in Fig. 2, the strong peak at around 3410 cm−1 was assigned to the absorption of hydroxyl groups in cellulose. Compared with FTIR result of MCC, a new band at about 1750 cm−1 was observed in each grafted copolymer spectrum, which was assigned to the carbonyl group. The methyl asymmetric deformation in PLA side chains at around 1475 cm−1, and the symmetric C–O–C stretching modes in ester groups at around 1200 cm−1 also appeared in the spectra of the copolymers.49 The results clearly proved that PLA chains had been grafted onto cellulose backbone by melt copolycondensation of LA with hydroxyl groups in cellulose and MCC-g-PLA copolymers were synthesized successfully, as shown in Scheme 1.
 | ||
| Fig. 2 FTIR spectra of (a) MCC, (b) MP-C0W4-T130, (c) MP-C0W4-T140, (d) MP-C0W4-T150, and (e) MP-C0W4-T160. | ||
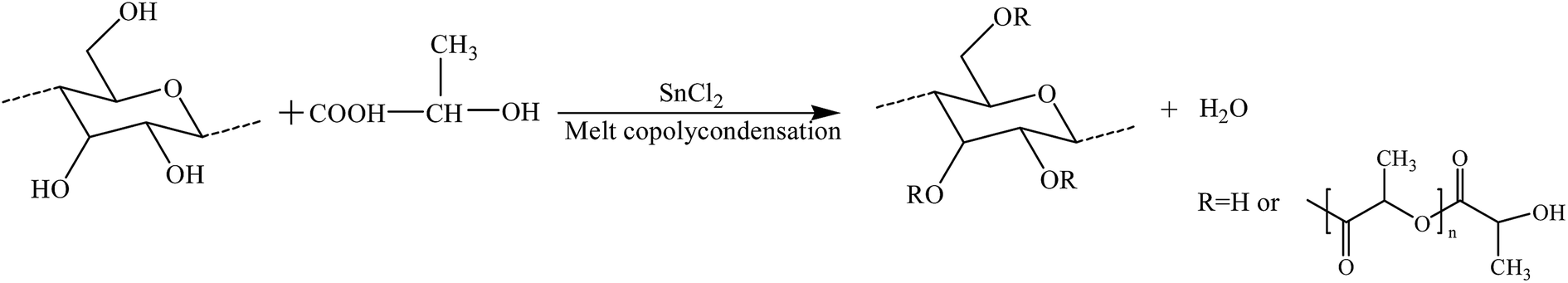 | ||
| Scheme 1 Illustration of the melt copolycondensation of LA with MCC. | ||
The structure of the copolymers was also investigated by 1H NMR. An 1H NMR spectrum obtained for one MCC-g-PLA copolymer (MP-C0W4-T160) was displayed in Fig. 3 and the signal peaks were assigned as follows: δ = 1.32 ppm (methyl protons of terminal lactyl); δ = 1.50 ppm (methyl protons of internal lactyl); δ = 4.23 ppm (methine protons of internal lactyl); δ = 5.07–5.29 ppm (methine protons of terminal lactyl); δ = 3–5.4 ppm (protons of anhydroglucose). The appearance of proton signals of lactyl groups in this spectrum, further confirmed that MCC-g-PLA copolymers were synthesized successfully.
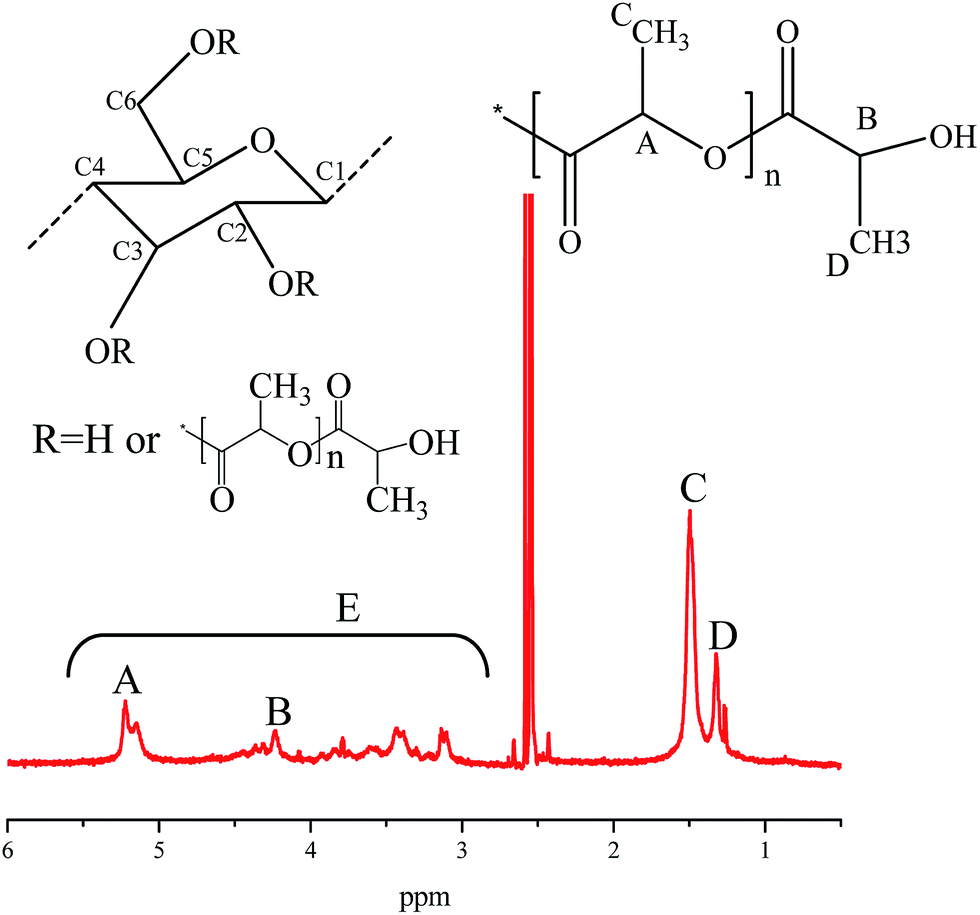 | ||
| Fig. 3 1H NMR spectrum of a MCC-g-PLA copolymer (MP-C0W4-T160). | ||
The contents of grafted PLA in MCC-g-PLA copolymers were calculated according to 1H NMR spectra. As shown in Fig. 3, we designated the resonance peak area derived from methine protons of internal lactyls in PLA side chains as A, and the resonance peak area from methine protons of terminal lactyls as B. The resonance peak area derived from methyl protons of internal lactyls was labeled as C, and the area from methyl protons of terminal lactyls was labeled as D. The integral of all protons of anhydroglucose (AHG) unit was designated as E. Then the average degree of polymerization of a PLA side chain (DPs), the degree of lactyl substitution (DS, the average number of hydroxyl groups substituted for lactyls per AHG of cellulose), and the molar substitution (MS, the average number of introduced lactyl units per AHG of cellulose) were estimated by eqn (1)–(3),38,40 respectively, and the results were summarized in Table 1.
| DPs = (IC + ID)/ID | (1) |
| DS = 7ID/3[IE − (IC + ID)/3] | (2) |
| MS = DPs × DS | (3) |
According to Table 1, the amount and length of PLA side chains can be controlled by adjusting reaction conditions and molar ratios of LA monomer to MCC, and the relationship between them will be discussed in detail subsequently.
From Table 1, the difference of the reaction conditions of samples MP-C0W4-TX (X = 130, 140, 150, 160) was reaction temperature, where the average numbers of hydroxyl groups substituted for lactyls per AHG (DS values) were almost constant, ranging from 0.60 to 0.67 with increasing reaction temperature, meaning that the accessibility of LA monomer to cellulose kept the same. Based on previous analysis, the accessibility of cellulose was mainly affected by solvent exchange pretreatment process. So the influence of reaction temperature on DS was small. However DPs values of the four samples increased with the increasing of reaction temperature. Lactic acid melting polycondensation reaction was an equilibrium reaction, producing PLA and water, so removing water was beneficial to the conversion of lactic acid into PLA.50,51 Under the same vacuum condition, high temperature could accelerate the removal of water, including free water and that formed in reaction process. Besides, elevated temperature could also increase the thermal degradation rate. Therefore, the influence of temperature on reaction was a combination of these factors. In the temperature range of our experiments, the DPs value increased with the temperature rising.
Samples MP-CXW4-T160 (X = 0, 1, 2) were synthesized by adjusting the molar ratios of LA monomer to MCC. According to Table 1, with increasing molar ratios of LA/AHG, DS and DPs values of the copolymers showed no obvious changes, indicating that increasing the content of LA monomer had almost no effect on the copolymers. Theoretically, increasing the content of reactant was helpful to produce the product for an equilibrium reaction. But in our work, the melt copolycondensation happened in a heterogeneous condition, and the steric hindrance influenced the reaction, which made the added monomer not participate in the reaction completely, so the influence of monomer content on graft copolymers was not so significant.
Except for the reaction process, the pretreatment process, in which we used solvent exchanged method to change the accessibility of cellulose, also influenced the formation of copolymers. The solvent exchanged process was related to the washing step with lactic acid in the pretreatment process, then we wondered whether the washing times with lactic acid had influence on the graft copolymers. So we synthesized samples MP-C2WX-T160 (X = 1, 2, 3, 4) through the same reaction process, only changing the washing times with lactic acid in pretreatment process.
From Table 1, we observed the dependence of DS and DPs on washing times. Obviously, DS and DPs increased gradually when the number of washing times reduced, indicating that the consumption of lactic acid can be reduced. During the process that the dissolved cellulose was regenerated and washed with lactic acid, firstly, lactic acid entered into cellulose, and combined with alkali, then the alkali was brought out of cellulose, at this time hydroxyl groups surrounded by alkali were freed out. These free hydroxyl groups probably formed hydrogen bonds with other free hydroxyl groups, or probably were surrounded by lactic acid again. If hydroxyl group contacted with lactic acid, it had the possibility of reacting with LA. But, once it formed hydrogen bond with other free hydrogen group, it was hard to take part in a reaction because the hydrogen bonds were difficult to be broken again. So in each washing process, some hydroxyl groups which were surrounded with alkali or lactic acid were freed out again and then formed hydrogen bonds, thus losing the possibility of reaction. So with the increase of washing times, DS value decreased gradually. The hydrogen bonds also limited the diffusion of lactic acid in cellulose, which affected the amount of lactic acid that could participate in reaction, causing the decrease of DPs.
The crystalline structures of MCC and MP-C2W1-T160 were studied by WAXD and the results were shown in Fig. 4 (Spectra of other copolymers were similar to MP-C2W1-T160 and not shown here for brevity). MCC was a typical cellulose I, as mentioned above. Different from WAXD spectrum of MCC, MP-C2W1-T160 showed two broad and small peaks at 19.8°and 22.0°, which were the characteristic diffraction peaks of cellulose II. The intensity of these two peaks was very weak, and the characteristic peak at 12.1° did not appear, indicating that MCC-g-PLA copolymers presented an imperfect crystalline structure of cellulose II. According to Kolpak and Blackwell,44 for cellulose II intramolecular hydrogen bonds (O2′–H⋯O6) and intermolecular hydrogen bonds (O6–H⋯O3, O6–H⋯O2) were in the 020 plane, and intermolecular hydrogen bonds (O2–H⋯O2′) were in the 110 plane. These hydrogen bonds made the molecular chains arrange regularly and form a perfect crystal. Reaction between LA and hydroxyl groups reduced the amount of hydroxyl groups capable of forming hydrogen bonds. On the other hand, the existence of PLA side chains generated steric hindrance, also affecting the formation of hydrogen bonds and regular arrangement of molecular chains. So the hydrogen-bonding network was weakened and MCC-g-PLA copolymers could not form perfect crystals. These results also proved that copolycondensation reaction occurred both in amorphous and crystalline region. Therefore we had improved the reactivity of cellulose by this method and synthesized MCC-g-PLA successfully.
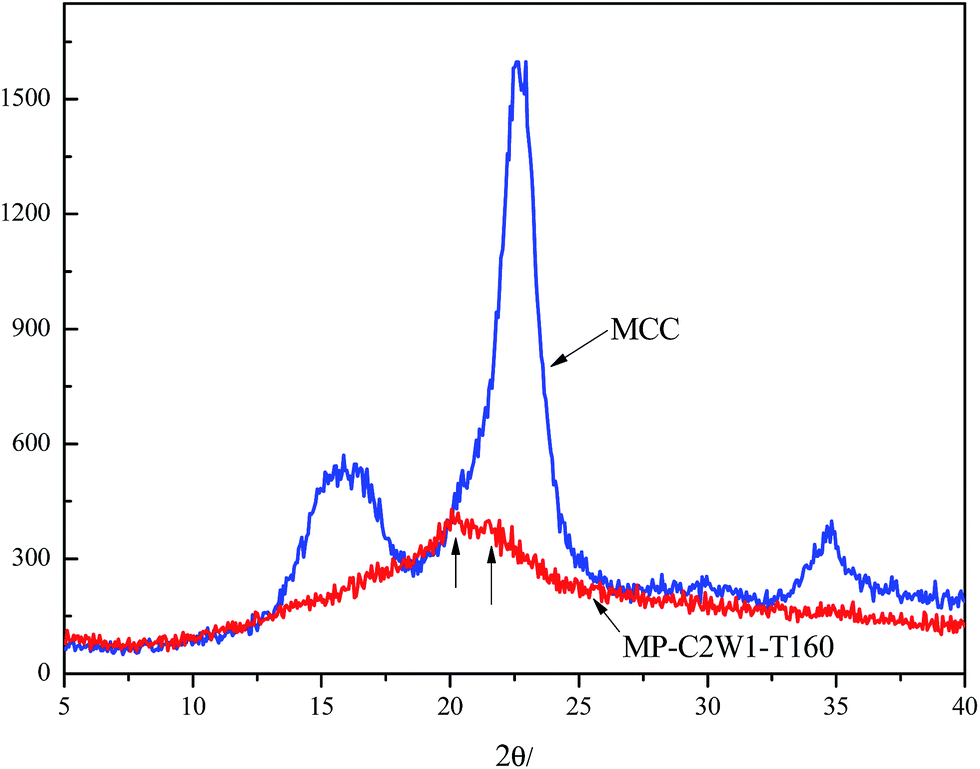 | ||
| Fig. 4 WAXD spectra of unmodified MCC and MP-C2W1-T160. | ||
3.2. Thermal properties of MCC-g-PLA copolymers
The thermal properties of MCC-g-PLA copolymers were investigated by DSC and TG measurements. Fig. 5 displayed the DSC curves of MCC and MCC-g-PLA copolymers (MP-C0W4-T130, MP-C0W4-T160, MP-C2W1-T160) in the second heating scan, and Tg values collected from second heating curves for all samples were listed in Table 2. From Fig. 5, each curve of MCC-g-PLA copolymers exhibited a baseline gap which reflected the glass transition temperature Tg. However in the standard heating scan unmodified cellulose did not show a glass transition temperature, Batzer and Kreibich52 reported a Tg at about 230 °C for cellulose by using a more sensitive method. It can be seen that Tg varied with the copolymer composition, Tg of MP-C0W4-T130 was the highest, and Tg of MP-C2W1-T160 was the lowest. Compared to the unmodified MCC, the remarkable decrease in Tg for MCC-g-PLA copolymers suggested that the existence of PLA side chains broke the hydrogen-bonding network, expanded the intermolecular distance, and improved the chain mobility. This was also confirmed by WAXD result. That is, the grafted PLA side chains played an important role of internal plasticizer to the semi-rigid cellulose. | ||
| Fig. 5 DSC curves of (a) MCC, (b) MP-C0W4-T130, (c) MP-C0W4-T160, and (d) MP-C2W1-T160. | ||
| Sample | DPs | DS | MS | Tg (°C) | Tonset (°C) | Tmax (°C) |
|---|---|---|---|---|---|---|
| MP-C0W4-T130 | 2.79 | 0.60 | 1.67 | 77.50 | 248.1 | 317.8 |
| MP-C0W4-T140 | 3.40 | 0.60 | 2.04 | 70.50 | 244.2 | 315.8 |
| MP-C0W4-T150 | 3.47 | 0.64 | 2.22 | 70.50 | 253.7 | 312.9 |
| MP-C0W4-T160 | 4.67 | 0.67 | 3.13 | 60.93 | 264.7 | 316.7 |
| MP-C1W4-T160 | 5.17 | 0.71 | 3.67 | 58.56 | 257.7 | 321.5 |
| MP-C2W4-T160 | 4.60 | 0.64 | 2.94 | 61.94 | 255.1 | 321.9 |
| MP-C2W3-T160 | 4.70 | 0.88 | 4.14 | 62.79 | 242.3 | 285.7, 321.6 |
| MP-C2W2-T160 | 5.90 | 0.88 | 5.19 | 59.99 | 239.4 | 277.1, 313.2 |
| MP-C2W1-T160 | 6.22 | 0.96 | 5.97 | 57.42 | 216.4 | 256.7, 301.7 |
| MCC | 320.1 | 347.6 |
TG measurement was used to evaluate the thermal stability of copolymers and the results were displayed in Fig. 6 and Table 2. Obviously, the onset decomposition temperature (Tonset) and the maximum decomposition temperature (Tmax) of MCC-g-PLA copolymers were lower than those of MCC. It was owing to that the introduction of PLA side chains on the cellulose backbone destroyed the hydrogen-bonding network and crystalline structure of MCC to some extent, resulting in the decrease of thermal stability. This was also confirmed by DSC and WAXD results. On the derivative thermogravimetry (DTG) curves, there was only one peak for copolymers when MS < 4.14, demonstrating that the thermal degradation of copolymers proceeded in one step. Because the content of PLA side chains was low, the decomposition peak of PLA chains did not emerge, and the peak in DTG curve was mainly caused by decomposition of cellulose backbone. However a shoulder peak appeared near the main peak in DTC curves of MP-C1W4-T160 and MP-C2W4-T160, showing that thermal decomposition process changed gradually. When MS ≥ 4.14, two derivate thermogravimetry peaks appeared, indicating that thermal degradation of these samples proceeded in two steps. This should be attributed to the different decomposition temperatures of MCC backbone and PLA side chains, and the degradation occurring at low temperature was caused by PLA side chains, and that at high temperature was caused by cellulose backbone, which was also reported in literature.40
 | ||
| Fig. 6 (a) TG and (b) DTG curves of MCC, MP-C0W4-T160, MP-C2W4-T160 and MP-C2W1-T160. | ||
3.3. Structure and properties of PLA/MCC-g-PLA composites
According to the above analysis, PLA had been successfully grafted onto the MCC, and then sample MP-C2W1-T160, which had the longest PLA side chains, was chosen and compounded into PLA matrix to explore the influence of MCC-g-PLA copolymer on the structure and properties of PLA. Improving the dispersion of cellulose in matrix is necessary to enhance the properties of such composites,53 and the dispersion state of MCC-g-PLA in PLA matrix was revealed by SEM micrograph shown in Fig. 7. For neat PLA, the micrograph showed a smooth and flat fracture surface. But it was changed to an uneven fracture surface with the addition of MCC-g-PLA, which indicated that a signification PLA matrix deformation occurred. As is known, the compatibility of PLA and cellulose was poor due to the hydrophobic nature of PLA and hydrophilic characteristic of cellulose.38,54 So large-size aggregates of cellulose and void appeared in the fracture surfaces of PLA/MCC composites, which was indicated with arrows in Fig. 7(e)–(g). However, MCC-g-PLA dispersed well in the matrix, and there was no obvious large-size agglomeration, showing that the existence of PLA side chains can greatly improve the dispersion state of MCC-g-PLA in PLA matrix and inhibit the agglomeration of MCC-g-PLA and improve the compatibility with PLA. | ||
| Fig. 7 SEM micrographs of (a) PLA, (b) PLA-MP-2, (c) PLA-MP-3, (d) PLA-MP-4, (e) PLA-MCC-2, (f) PLA-MCC-3, and (g) PLA-MCC-4. | ||
In order to investigate the influence of MCC-g-PLA on the melt strength of PLA, the extensional rheological properties of PLA and PLA/MCC-g-PLA composites were characterized by the extensional rheology at the extensional strain rates of 0.1 s−1 and 0.5 s−1 in steady uniaxial extension, and the results were shown in Fig. 8. It was obvious that the elongational viscosity of neat PLA kept almost constant at the full time of stretching, and the strain-hardening behavior could not be observed, which was the typical characteristic of linear polymer. The elongational viscosity was improved with the addition of MCC-g-PLA at low extensional strain rate (0.1 s−1), and the increment increased with increasing content of MCC-g-PLA, which was attributed to the entanglement between MCC-g-PLA and PLA matrix. According to the 1H NMR analysis of MCC-g-PLA, the average degree of polymerization of PLA side chain was 6.22, that is the average molecular weight was 425 g mol−1, however the critical molecular weight for entanglement of PLA was near 9000 g mol−1,55 the length of PLA side chains in MCC-g-PLA was not long enough to form strong entanglement with PLA matrix. So the entanglement should be between MCC-g-PLA filler particles and PLA matrix, but not between the PLA side chain and PLA matrix. However, at high extensional strain rate (0.5 s−1), when the content of filler was low (sample PLA-MP-2 and PLA-MP-3), the elongational viscosity could not be improved and was approximately equal to that of neat PLA; only PLA-MP-4 showed higher elongational viscosity than neat PLA. According to the SEM results, MCC-g-PLA was well dispersed in the matrix, so the higher the content of MCC-g-PLA, the more the entanglement in the composites. For PLA-MP-2, due to the low entanglement, the elongational viscosity was almost equal to neat PLA even at low extensional strain rate. For PLA-MP-3, the entanglement was increased, so the elongational viscosity increased at low extensional strain rate. But at high extensional strain rate, the elongational viscosity decreased because of disentanglement and was approximately equal to neat PLA indicating hardly any entanglement between filler and matrix. For PLA-MP-4, the elongational viscosity decreased with the increase of extensional strain rate, but was still higher than neat PLA, showing that there was entanglement remained in spite of the existence of disentanglement.
 | ||
| Fig. 8 Variation of elongation viscosity as a function of time for PLA and PLA/MCC-g-PLA composites at 158.5 °C with elongation rates of (a) 0.1 s−1 and (b) 0.5 s−1. | ||
The effects of MCC-g-PLA on the non-isothermal crystallization and melting behaviors of PLA matrix were also investigated by DSC. Cooling curves of different samples were shown in Fig. 9(a). For pure PLA, there was no exothermic peak representing the crystallization of PLA in the cooling curve due to the relatively rigid molecular chain and slow crystallization rate. However, an exothermic peak emerged when MCC-g-PLA was introduced, and the peak became stronger with increasing content of MCC-g-PLA, except for sample PLA-MP-4. In the second heating process (Fig. 9(b)), there was no cold crystallization peak, which was also observed in literature,56,57 and melting peak for pure PLA. For PLA/MCC-g-PLA composites, we observed the appearance of cold crystallization peak, and the cold crystallization temperature (TCC) shifted to the lower temperature with increasing content of MCC-g-PLA, and returned to higher temperature for PLA-MP-4. These results indicated that the addition of MCC-g-PLA acted as nucleation agent and promoted the crystallization ability of PLA. At first, the nucleation ability of MCC-g-PLA was enhanced with the increasing content of nucleation agent, thus making crystallization peak become stronger and TCC shift to lower temperature. Then the content of MCC-g-PLA increased further, the entanglement between PLA and MCC-g-PLA also increased, resulting in the hindered mobility of PLA molecular chains. This was in agreement with the extensional rheology test. In addition, the shape of melting peak also changed with the content of MCC-g-PLA. As shown in Fig. 9(b), there were double melting peaks in the second heating curves of PLA/MCC-g-PLA composites, which was attributed to the melt/recrystallization behavior of PLA.58,59 The melting peak at lower temperature was caused by the melting of imperfect crystals and the melting peak at higher temperature was caused by the melting of the relatively perfect crystal and recrystallized crystals. Taking PLA-MP-3 as an example, the crystallization ability was the strongest, and the amount of relatively perfect crystal formed in crystallization process was more, so the higher temperature melting peak was intensified.
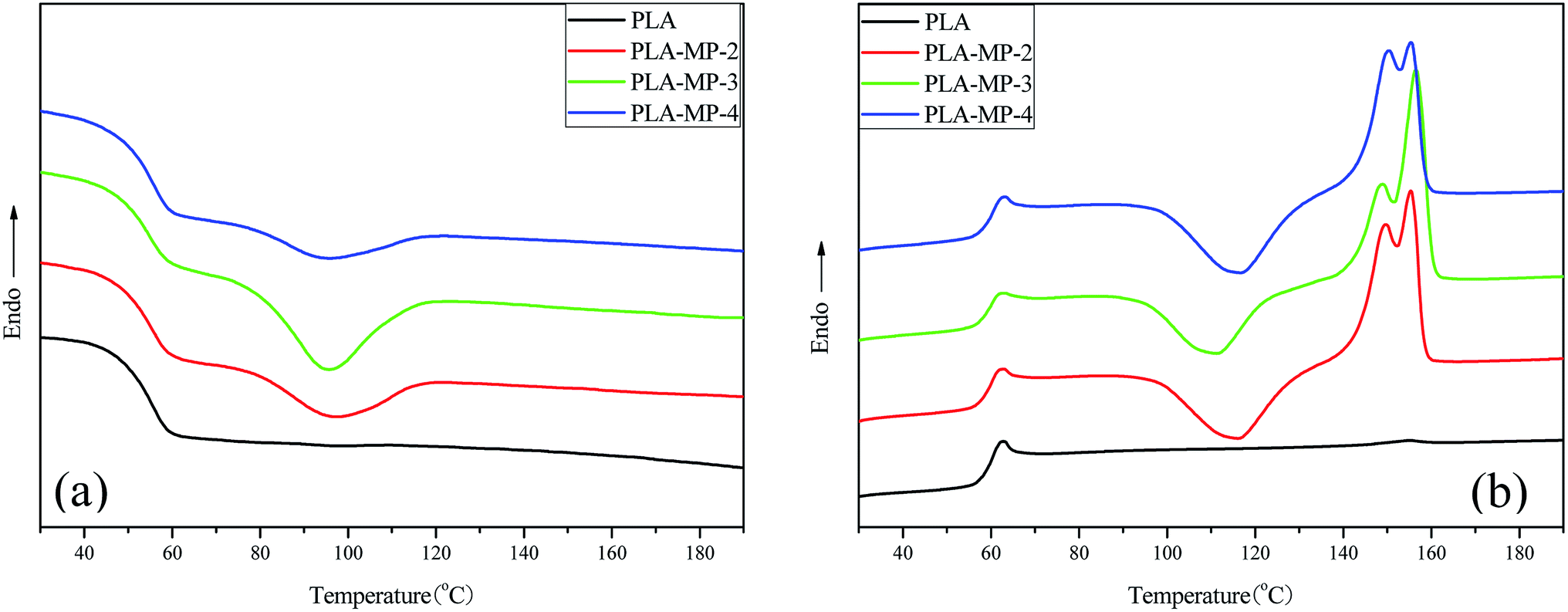 | ||
| Fig. 9 DSC thermograms of pure PLA and PLA/MCC-g-PLA composites (a) for the first cooling process, (b) for the second heating process. | ||
4. Conclusions
A series of cellulose derivatives MCC-g-PLA copolymers with MS of PLA in a range of 1.67–5.97 were synthesized by melt copolycondensation of lactic acid with microcrystalline cellulose using SnCl2 as catalyst. The factors of reaction temperature and washing times in pretreatment process had a great effect on the DS and DPs value of graft copolymers. The resultant copolymers had an imperfect crystalline structure of cellulose II due to the PLA side chains, which was confirmed by WAXD measurements. All MCC-g-PLA copolymers exhibited a single Tg in second heating thermograms in DSC, and Tg varied from 77.50 °C to 57.77 °C in relation to the MS of PLA, indicating that the existence of PLA side chains had played an important role as internal plasticization. The thermal stability of MCC-g-PLA copolymer was lower than that of MCC, due to the weakened hydrogen-bonding network and crystallinity. When MS ≥ 4.14, the thermal degradation of copolymers proceeded in two steps instead of one step, and the thermal decomposition behavior of PLA side chains appeared.MCC-g-PLA copolymer can be well dispersed in PLA matrix, and the melt strength of PLA can be improved by the addition of MCC-g-PLA at low extensional strain rate, but the improvement decreased at high extensional strain rate, due to the weak entanglement. MCC-g-PLA also acted as nucleation agent to enhance the crystallization ability of PLA. The results in this work proved that the performance of PLA can be improved by this method, but the length of PLA side chains was short, if we improve its length over the critical entanglement length, the PLA side chain can also form entanglement with matrix, thus making melt strength increase significantly, so the studies on further improving the length of PLA side chains are now in progress.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NNSFC Grant 51421061) and the Major State Basic Research Development Program of China (973 program) (2011CB606006).References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- A. K. Mohanty, M. Misra and G. Hinrichsen, Macromol. Mater. Eng., 2000, 276–277, 1–24 CrossRef.
- M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC.
- Y. Zhao, Z. Wang, J. Wang, H. Mai, B. Yan and F. Yang, J. Appl. Polym. Sci., 2004, 91, 2143–2150 CrossRef CAS.
- T. Kaito, A. Myoui, K. Takaoka, N. Saito, M. Nishikawa, N. Tamai, H. Ohgushi and H. Yoshikawa, Biomaterials, 2005, 26, 73–79 CrossRef CAS PubMed.
- H. S. Na and S. C. Kim, J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 254–264 CrossRef CAS.
- F. Ge, Y. Ding, L. Yang, Y. Huang, L. Jiang and Y. Dan, RSC Adv., 2015, 5, 70473–70481 RSC.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- D. Howard, K. Partridge, X. Yang, N. M. P. Clarke, Y. Okubo, K. Bessho, S. M. Howdle, K. M. Shakesheff and R. O. C. Oreffo, Biochem. Biophys. Res. Commun., 2002, 299, 208–215 CrossRef CAS PubMed.
- P. Zhang, Z. Hong, T. Yu, X. Chen and X. Jing, Biomaterials, 2009, 30, 58–70 CrossRef CAS PubMed.
- H. Shi, Q. Gan, X. Liu, Y. Ma, J. Hu, Y. Yuan and C. Liu, RSC Adv., 2015, 5, 79703–79714 RSC.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- I. Kyrikou and D. Briassoulis, J. Polym. Environ., 2007, 15, 125–150 CrossRef CAS.
- R. Bhardwaj and A. K. Mohanty, Biomacromolecules, 2007, 8, 2476–2484 CrossRef CAS PubMed.
- H. Li and M. A. Huneault, Polymer, 2007, 48, 6855–6866 CrossRef CAS.
- F. Wu, B. Zhang, W. Yang, Z. Liu and M. Yang, Polymer, 2014, 55, 5760–5772 CrossRef CAS.
- M. Beaulne and E. Mitsoulis, J. Appl. Polym. Sci., 2007, 105, 2098–2112 CrossRef CAS.
- X. Lan, X. Li, Z. Liu, Z. He, W. Yang and M. Yang, J. Macromol. Sci., Part A: Pure Appl. Chem., 2013, 50, 861–870 CrossRef CAS.
- Y. Di, S. Iannace, E. Di Maio and L. Nicolais, Macromol. Mater. Eng., 2005, 290, 1083–1090 CrossRef CAS.
- R. Mehta, V. Kumar, H. Bhunia and S. N. Upadhyay, J. Macromol. Sci., Part C: Polym. Rev., 2005, 45, 325–349 CrossRef.
- H. J. Lehermeier and J. R. Dorgan, Polym. Eng. Sci., 2001, 41, 2172–2184 CAS.
- L. Wang, X. Jing, H. Cheng, X. Hu, L. Yang and Y. Huang, Ind. Eng. Chem. Res., 2012, 51, 10088–10099 CrossRef CAS.
- D. Auhl, J. Stange, H. Münstedt, B. Krause, D. Voigt, A. Lederer, U. Lappan and K. Lunkwitz, Macromolecules, 2004, 37, 9465–9472 CrossRef CAS.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- H. Lönnberg, Q. Zhou, H. Brumer, T. T. Teeri, E. Malmström and A. Hult, Biomacromolecules, 2006, 7, 2178–2185 CrossRef PubMed.
- W. Yuan, J. Yuan, F. Zhang and X. Xie, Biomacromolecules, 2007, 8, 1101–1108 CrossRef CAS PubMed.
- M. Krouit, J. Bras and M. N. Belgacem, Eur. Polym. J., 2008, 44, 4074–4081 CrossRef CAS.
- Y. Teramoto and Y. Nishio, Polymer, 2003, 44, 2701–2709 CrossRef CAS.
- X. Samain, V. Langlois, E. Renard and G. Lorang, J. Appl. Polym. Sci., 2011, 121, 1183–1192 CrossRef CAS.
- X. Zhang, J. Zhao, L. Cheng, C. Lu, Y. Wang, X. He and W. Zhang, RSC Adv., 2014, 4, 55195–55201 RSC.
- A. Jayalakshmi, I.-C. Kim and Y.-N. Kwon, RSC Adv., 2015, 5, 48290–48300 RSC.
- M. Ioelovich, BioResources, 2009, 4, 1168–1177 CAS.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- T. Röder, B. Morgenstern, N. Schelosky and O. Glatter, Polymer, 2001, 42, 6765–6773 CrossRef.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- A. Mayumi, T. Kitaoka and H. Wariishi, J. Appl. Polym. Sci., 2006, 102, 4358–4364 CrossRef CAS.
- H. Dong, Q. Xu, Y. Li, S. Mo, S. Cai and L. Liu, Colloids Surf., B, 2008, 66, 26–33 CrossRef CAS PubMed.
- S. Xiao, T. Yuan, H. Cao, D. Lin, Y. Shen, J. He and B. Wang, BioResources, 2012, 7, 1748–1759 CrossRef.
- C. Yan, J. Zhang, Y. Lv, J. Yu, J. Wu, J. Zhang and J. He, Biomacromolecules, 2009, 10, 2013–2018 CrossRef CAS PubMed.
- L. Dai, D. Li and J. He, J. Appl. Polym. Sci., 2013, 130, 2257–2264 CrossRef CAS.
- Y. Zhang, X. Li, Y. Yang, A. Lan, X. He and M. Yu, RSC Adv., 2014, 4, 34584–34590 RSC.
- A. Isogai, M. Usuda, T. Kato, T. Uryu and R. H. Atalla, Macromolecules, 1989, 22, 3168–3172 CrossRef CAS.
- F. J. Kolpak and J. Blackwell, Macromolecules, 1976, 9, 273–278 CrossRef CAS PubMed.
- P. Langan, Y. Nishiyama and H. Chanzy, Biomacromolecules, 2001, 2, 410–416 CrossRef CAS PubMed.
- P. K. Gupta, V. Uniyal and S. Naithani, Carbohydr. Polym., 2013, 94, 843–849 CrossRef CAS PubMed.
- A. Isogai and R. H. Atalla, Cellulose, 1998, 5, 309–319 CrossRef CAS.
- M. G. Porter and R. S. Murray, Grass Forage Sci., 2001, 56, 405–411 CrossRef CAS.
- Y. Luan, J. Wu, M. Zhan, J. Zhang, J. Zhang and J. He, Cellulose, 2013, 20, 327–337 CrossRef CAS.
- E. M. Filachione and C. H. Fisher, Ind. Eng. Chem., 1944, 36, 223–228 CrossRef CAS.
- S. I. Moon, C. W. Lee, M. Miyamoto and Y. Kimura, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1673–1679 CrossRef CAS.
- H. Batzer and U. Kreibich, Polym. Bull., 1981, 5, 585–590 CrossRef CAS.
- B. Braun and J. R. Dorgan, Biomacromolecules, 2009, 10, 334–341 CrossRef CAS PubMed.
- E. Espino-Pérez, J. Bras, V. Ducruet, A. Guinault, A. Dufresne and S. Domenek, Eur. Polym. J., 2013, 49, 3144–3154 CrossRef.
- J. R. Dorgan, J. Janzen, M. P. Clayton, S. B. Hait and D. M. Knauss, J. Rheol., 2005, 49, 607–619 CrossRef CAS.
- J. M. Raquez, Y. Murena, A. L. Goffin, Y. Habibi, B. Ruelle, F. DeBuyl and P. Dubois, Compos. Sci. Technol., 2012, 72, 544–549 CrossRef CAS.
- E. Fortunati, I. Armentano, Q. Zhou, D. Puglia, A. Terenzi, L. A. Berglund and J. M. Kenny, Polym. Degrad. Stab., 2012, 97, 2027–2036 CrossRef CAS.
- T. Dobreva, J. M. Pereña, E. Pérez, R. Benavente and M. García, Polym. Compos., 2010, 31, 974–984 CrossRef CAS.
- Y. Song, K. Tashiro, D. Xu, J. Liu and Y. Bin, Polymer, 2013, 54, 3417–3425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |