DOI:
10.1039/C5RA23697E
(Paper)
RSC Adv., 2016,
6, 1143-1150
Thiacalix[4]arene-supported heterodinuclear NiII–LnIII complexes: slow magnetic relaxation behavior in the dysprosium analogue†
Received
10th November 2015
, Accepted 14th December 2015
First published on 17th December 2015
Abstract
Three heterodinuclear complexes, [(NiL1)Ln(L2)(CH3OH)]·acetone (Ln = Gd (1), Tb (2), Dy (3); H3L1 = 1,1,1-tris[(salicylideneamino)methyl]ethane), were stepwise synthesized based on a thiacalix[4]arene ligand (H2L2 = 5,11,17,23-tetrakis(1,1-dimethylethyl)-25,27-dihydroxy-26,28-dimethoxy thiacalix[4]arene). In 1–3, NiII and LnIII ions are doubly bridged by two phenoxo O atoms of L1. The NiII ion, only coordinated by the L1 ligand, features a {NiN3O3} coordination sphere; and the LnIII ion adopts a seven-coordinated {LnO7} environment, capped by one thiacalix[4]arene ligand in a bowl-shaped conformation. Magnetic studies on all complexes reveal that ferromagnetic couplings are operative between the NiII and LnIII ions. The dysprosium analogue (3) exhibits field-induced single-molecule magnet (SMM) behavior under a dc-applied field of 800 Oe. Moreover, the Dy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y (1:10) magnetically diluted sample (3′) shows slow magnetic relaxation at zero dc field. The results demonstrate that the stepwise synthetic method is more useful and effective than self-assembly for designing new 3d–4f heterodinuclear complexes with the thiacalix[4]arene ligand or other derivatives. The introduction of this type of thiacalix[4]arene ligand into the heterodinuclear complexes renders them promising candidates for new SMMs.
:Y (1:10) magnetically diluted sample (3′) shows slow magnetic relaxation at zero dc field. The results demonstrate that the stepwise synthetic method is more useful and effective than self-assembly for designing new 3d–4f heterodinuclear complexes with the thiacalix[4]arene ligand or other derivatives. The introduction of this type of thiacalix[4]arene ligand into the heterodinuclear complexes renders them promising candidates for new SMMs.
Introduction
In the past decades, single-molecule magnets (SMMs)/single-chain magnets (SCMs) have attracted much attention from chemists owing to their potential applications for high-density information storage, quantum computing, and molecular spintronics.1 Since the first 3d–4f heterodinuclear SMM was reported in 2004,2 continued efforts have been focused on the combination of 3d and 4f metal ions for new polynuclear clusters.3 Some 3d transition metal ions, especially the NiII ion with a relatively large high-spin ground-state, often exhibit a large single-ion zero-field splitting.4 4f ions, such as DyIII, TbIII, usually exhibit large ground-state spin and large intrinsic magnetic anisotropy.5 The ferromagnetic coupling between 3d and 4f ions often results in a high-spin ground state and may endow SMMs with a high energy barrier. A number of 3d–4f SMMs/SCMs have been reported to be ferromagnetic couplings in the literature,6 such as [Dy2IIINiII(Sa)8]·(HSa)2 (HSa = salicylic aldehyde) reported by Tang et al., of which the Ueff is 55.19 K.6c
To the best of our knowledge, the structure of the selected ligand plays the crucial role in forming novel frameworks and tuning magnetic properties.7 The Schiff bases are very popular ligands serving to construct 3d–4f complexes.6a,8 Take the tripodal Schiff base (1,1,1-tris[(salicylideneamino)methyl]ethane, H3L1) as an example, this type of ligand has two distinct bonding sites with the inner compartment (N3O3) to chelate 3d metal ion and the outer to accommodate 4f lanthanide ion.9 Yamaguchi et al. reported a NiII-complex, [Ni(H1.5L1)]2Cl·2CH3OH, based on the tripodal ligand H3L1,9a in which the NiII ion is chelated by a N3O3 group of H3L1. Whereafter, [(NiIIL1)DyIII(hfac)2(EtOH)] (Hhfac = hexafluoroacetylacetone) was synthesized. The outer O atoms coordinate 4f ions to afford 3d–4f heterodinuclear complex. However, the out-of-phase signal (χ′′) of [(NiIIL1)DyIII(hfac)2(EtOH)] was quite weak.9b In our previous work, we have reported a series of calix[4]arene-supported lanthanide complexes, in which the DyIII analogue shows obvious SMM behavior.10a Calix[4]arene/thiacalix[4]arene derivatives with rigid conformation,10 are well known to be good ligand candidates for the construction of lanthanide SMMs.10b,10c Usually, thiacalix[4]arene and its derivatives tend to produce clusters due to their multidentate coordination sites.11 Thus, the synthesis of mononuclear or 3d–4f dinuclear complexes based on thiacalix[4]arene is still a challenging research, which is rarely reported. To further investigate the advantages of thiacalix[4]arene in building SMMs, we aim to introduce it into 3d–4f heterodinuclear systems by the stepwise approach12 in order to control the organization process and study their magnetic properties.
In this paper, three isomorphous NiII–LnIII heterodinuclear complexes, [(NiL1)Ln(L2)(CH3OH)]·acetone (Ln = Gd (1); Ln = Tb (2); Ln = Dy (3); H3L1 = 1,1,1-tris[(salicylideneamino)methyl]ethane; H2L2 = 5,11,17,23-tetrakis(1,1-dimethylethyl)-25,27-dihydroxy-26,28-dimethoxy thiacalix[4]arene) (Scheme 1) were successfully synthesized by treating [Ni(H1.5L1)]2Cl·2CH3OH with Ln(acac)3·2H2O (Hacac = acetylacetone) and thiacalix[4]arene ligand stepwise. Herein, the syntheses, crystal structures and magnetic properties are presented. Complex 3 shows a field-induced SMM behavior. Interestingly, a Dy:Y (molar ratio of 1:10) magnetically diluted sample (3′) exhibits SMM behavior at zero applied direct-current (dc) field, which conforms the important role of the dipole–dipole interaction between magnetic centers in favoring the fast quantum tunneling of magnetization (QTM) process.
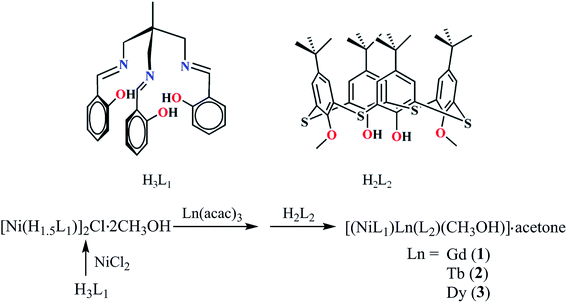 |
| | Scheme 1 Ligands used in this work and synthetic route of 1–3. | |
Experimental section
General information
All chemicals and solvents were obtained and used directly from the commercial sources. Starting materials, [Ni(H1.5L1)]2Cl·2CH3OH based on tripodal ligand 1,1,1-tris[(salicylideneamino)methyl]ethane (H3L1) and thiacalix[4]arene ligand (H2L2), were prepared according to the published literatures.9a,13 Elemental analyses were performed on a Elementar Vario MICRO analyzer. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 400–4000 cm−1 range using aVector22 Bruker spectrophotometer. XRD patterns were obtained on a D8 ADVANCE X-ray powder diffractometer (XRPD) with Cu Kα radiation (λ = 1.5405 Å). Inductively coupled plasma optical emission spectrometer (ICP-OES) analysis was carried out using a Perkin Elmer Optima 5300DV spectrometer. Magnetic susceptibility measurements for polycrystalline samples were performed on a Quantum Design MPMS-SQUID-VSM magnetometer in the temperature range from 2 to 300 K for direct current (dc) applied fields ranging from 0 to 70 kOe. Alternating current (ac) susceptibilities were obtained using an oscillating ac field of 2 Oe and in the frequency range 1–999 Hz. Diamagnetic corrections were calculated using Pascal's constants,14 and an experimental correction for the diamagnetic sample holder was applied.
Crystal structure determination
The crystal structures were determined on a Bruker Smart Apex II CCD-based diffractometer (Mo Kα radiation, λ = 0.71073 Å). The raw frame data were integrated into SHELX-format reflection files and corrected for Lorentz and polarization effects using SAINT.15 Corrections for incident and diffracted beam absorption effects were applied using SADABS supplied by Bruker.16 None of the crystals showed evidence of crystal decay during data collection. All structures were solved and refined against F2 by the full-matrix least-squares using the SHELXL-97 program.17 The positions of the metal atoms and their first coordination spheres were located from direct method E-maps. All non-hydrogen atoms were refined with anisotropic thermal parameters, and hydrogen atoms of the organic ligands and the hydroxide anion were calculated theoretically onto the specific atoms and refined isotropically with fixed thermal factors. More details for the crystal data, data collection parameters, and refinement statistics were given in Table 1. Relevant interatomic bond distances and bond angles were listed in Table 2 and S1,† respectively. CCDC reference numbers 1424163 (1), 1424164 (2), 1424165 (3).
Table 1 Crystal data and structure refinement for 1–3
| |
1 |
2 |
3 |
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]0.5. |
| Empirical formula |
C72H84GdN3NiO9S4 |
C72H84TbN3NiO9S4 |
C72H84DyN3NiO9S4 |
| Formula weight |
1479.62 |
1481.29 |
1484.87 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| a (Å) |
11.7413(8) |
11.7563(15) |
11.7432(11) |
| b (Å) |
17.4898(11) |
17.521(2) |
17.4859(17) |
| c (Å) |
18.9005(12) |
18.887(2) |
18.8642(18) |
| α (°) |
72.1160(10) |
72.230(2) |
72.2850(10) |
| β (°) |
79.3280 |
79.386(2) |
79.4140(10) |
| γ (°) |
77.7270 |
77.672(2) |
77.774(2) |
| V (Å3) |
3579.2(4) |
3589.5(8) |
3576.5(6) |
| ρ calc (g cm−3) |
1.373 |
1.371 |
1.379 |
| F (000) |
1530 |
1532 |
1534 |
| Data/restraints/parameters |
13136/0/829 |
13282/0/829 |
13242/0/829 |
| GOF on F2 |
1.110 |
1.027 |
1.063 |
| R1a/wR2b [I > 2σ(I)] |
0.0433/0.1227 |
0.0473/0.1226 |
0.0367/0.1032 |
| R1/wR2 [all data] |
0.0518/0.1426 |
0.0676/0.1487 |
0.0417/0.1094 |
Table 2 Selected bond lengths (Å) and Ln⋯Ni distances (Å) for 1–3
| |
1 (Gd) |
2 (Tb) |
3 (Dy) |
| Ln(1)–O(1) |
2.366(3) |
2.363(4) |
2.350(2) |
| Ln(1)–O(3) |
2.337(3) |
2.323(4) |
2.297(3) |
| Ln(1)–O(4) |
2.554(3) |
2.550(4) |
2.547(2) |
| Ln(1)–O(5) |
2.174(3) |
2.161(4) |
2.148(2) |
| Ln(1)–O(6) |
2.595(3) |
2.589(4) |
2.580(3) |
| Ln(1)–O(7) |
2.177(3) |
2.165(4) |
2.157(3) |
| Ln(1)–O(8) |
2.420(3) |
2.408(4) |
2.398(3) |
| Ni(1)–O(1) |
2.070(3) |
2.066(4) |
2.070(3) |
| Ni(1)–O(2) |
2.047(3) |
2.048(4) |
2.045(3) |
| Ni(1)–O(3) |
2.075(3) |
2.062(4) |
2.077(3) |
| Ni(1)–N(1) |
2.032(4) |
2.022(5) |
2.040(3) |
| Ni(1)–N(2) |
2.032(4) |
2.039(5) |
2.034(3) |
| Ni(1)–N(3) |
2.078(4) |
2.093(5) |
2.087(3) |
| Ln(1)⋯Ni(1) |
3.4712(6) |
3.4568(8) |
3.4489(6) |
Synthesis of [(NiL1)Gd(L2) (CH3OH)]·acetone (1). Taking complex [Ni(H1.5L1)]2Cl·2CH3OH (5.18 mg, 0.01 mmol) as the ligand to mixed with Et3N (2.02 mg, 0.02 mmol) in methanol (4 mL), resulting in a yellow solution. Then Gd(acac)3·2H2O (9.96 mg, 0.02 mmol) was added. After stirring for 30 min, the solution appeared turbid, followed by adding H2L2 (14.98 mg, 0.02 mmol) and acetone (4 mL). The reaction mixture was heated at 85 °C for 3 days, and then cooled to the room temperature. Greenish yellow block-shaped crystals of 1 were obtained. Yield, 70%. Elemental analysis (%) calculated for C72H84GdN3NiO9S4: C 58.44, H 5.72, S 8.67. Found: C, 58.69, H, 5.66, S 8.71. Selected IR frequencies (KBr pellet, cm−1): 3619(w), 2960(s), 1650(s), 1634(m), 1443(vs), 1415(m), 1313(m), 1259(w), 1125(w), 1024(m), 878(w), 795(m), 593(w).
Synthesis of [(NiL1)Tb(L2)(CH3OH)]·acetone (2). The procedure was similar to the synthesis of 1 except that Tb(acac)3·2H2O was used in place of Gd(acac)3·2H2O. Greenish yellow block-shaped crystals of 2 were obtained. Yield, 60%. Elemental analysis (%) calculated for C72H84TbN3NiO9S4: C 58.38, H 5.72, S 8.66. Found: C, 58.55, H, 5.63, S 8.60. Selected IR frequencies (KBr pellet, cm−1): 3443(w), 2960(m), 1634(s), 1551(m), 1443(vs), 1415(m), 1313(m), 1259(w), 1151(w), 1024(w), 845(m), 754(m), 592(w).
Synthesis of [(NiL1)Dy(L2)(CH3OH)]·acetone (3). The procedure was similar to the synthesis of 1 except that Dy(acac)3·2H2O was used in place of Gd(acac)3·2H2O. Greenish yellow block-shaped crystals of 3 were obtained. Yield, 65%. Elemental analysis (%) calculated for C72H84DyN3NiO9S4: C 58.24, H 5.70, S 8.64. Found: C, 58.58, H 5.79, S 8.55. Selected IR frequencies (KBr pellet, cm−1): 3446(m), 2960(m), 1634(s), 1442(vs), 1415(m), 1314(m), 1259(m), 1122(w), 1024(w), 879(m), 754(m), 593(w).
Synthesis of [(NiL1)Dy0.071Y0.929(L2) (CH3OH)]·acetone (3′). The diluted complex was prepared by following the same method as that for 1 but using Dy(acac)3·2H2O (0.895 mg, 0.0018 mmol) and Y(acac)3·2H2O (7.65 mg, 0.018 mmol) (Dy:Y = 1:10) instead of Dy(acac)3·2H2O (9.94 mg, 0.02 mmol). The final greenish yellow crystals of the doped complex were obtained with a yield of 51%. Elemental analysis (%) calculated for C72H84Dy0.071Y0.929N3NiO9S4: C 61.05, H 5.98, S 9.05. Found: C, 59.75, H 5.76, S 8.99. IR frequencies (KBr pellet, cm−1): 3444(w), 2960(m), 1650(s), 1634(m), 1443(vs), 1415(m), 1314(m), 1260(w), 1153(w), 1024(m), 878(w), 755(m), 593(w).
Results and discussion
Synthesis and characterizations
Complexes 1–3 were isolated by the stepwise synthetic approach in good yield (Scheme 1, ESI†). Firstly, [Ni(H1.5L1)]2Cl·2CH3OH and Ln(acac)3·2H2O were reacted in methanol, which became increasingly turbid after stirring. Then, H2L2 and acetone were added and the mixture was heated at 85 °C to obtain greenish yellow block-shaped crystals. The acac anion plays an important role in the stepwise synthetic process. It can coordinate to metal centers and occupy the coordination sites of LnIII ions, but will more easily be replaced by other O-containing ligands such as thiacalix[4]arene. The stepwise synthetic approach is useful and effective for new 3d–4f heterodinuclear complexes, and it also enriches the type of metal complexes achievable based on the thiacalix[4]arenes moiety.
Crystal structures
The X-ray crystal structure analyses reveal that 1–3 are isomorphous except for some differences in bond lengths and angles (Table 2 and Fig. S1, ESI†). All complexes crystallize in the triclinic P space group. Each of them is a face-sharing dinuclear molecule, containing one NiII–LnIII heterometallic unit (Fig. 1). The NiII ion lies in an approximately octahedral {NiN3O3} coordination sphere composed of three imine N atoms (Ni–N = 2.032(4)–2.078(4) Å for 1, 2.022(5)–2.093(5) Å for 2, and 2.034(3)–2.087(3) Å for 3, respectively) and three phenolic O atoms (Ni–O = 2.047(3)–2.075(3) Å for 1, 2.048(4)–2.066(4) Å for 2, and 2.045(3)–2.077(3) Å for 3, respectively) from one tripodal ligand L1. Every LnIII ion is seven-coordinated {LnO7} by four O atoms of L2, two O atoms of L1, and one methanol O atom. The average Ln–O distance is 2.375, 2.366 and 2.354 Å for 1, 2, and 3, respectively [in what follows, three values separated by slashes will mean a sequence in the order 1/2/3], which are within the normal range according to previous reported values.18 It is worth noting that one of the three phenolic O atoms from L1 is uncoordinated to a LnIII ion with a long Ln⋯O2 distance of 3.939/3.931/3.922 Å. Within the NiII–LnIII heterometallic unit, NiII and LnIII ions are bridged by two O atoms (O1 and O3) form L1 with the average Ln–O distance of 2.352/2.343/2.324 Å. The intramolecular Ni⋯Ln distance is 3.471/3.457/3.449 Å, comparable with those in the similar phenolic O atoms-bridged NiII–LnIII complexes.18,19 Subsequently, the average bond angle of Ni–O–Ln is 103.19/103.15/103.19, and hinge angle (dihedral angle between the O–Ni–O and O–Ln–O planes) is 2.776/2.689/2.978°.
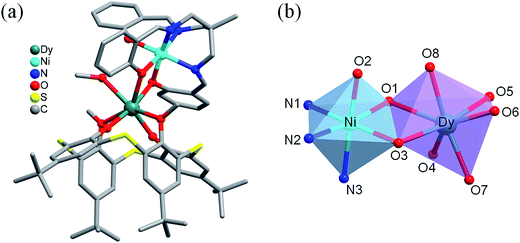 |
| | Fig. 1 (a) Crystal structure of complex [(NiL1)Dy(L2)(CH3OH)]·acetone (3). Hydrogen atoms and uncoordinated solvents are omitted for clarity. (b) Local coordination geometries of NiII and DyIII ions. | |
In the solid state, the acetone solvent molecule connects to the dinuclear unit via weak C–H⋯O and C–H⋯π interactions (Fig. S2a†), and two kinds of C–H⋯π interactions link the dinuclear units into a 1D chain with the shortest Ni⋯Ni distance of 7.998/8.021/8.023 Å and the shortest Ln⋯Ln distance of 9.673/9.702/9.698 Å (Fig. S2b and c†).
Thermogravimetric analyses and X-ray powder diffraction analyses
All of the complexes are stable under ambient conditions. Thermogravimetric analyses (TGA) of 1–3 were carried out from 30 to 900 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere. In 1–3, a weight loss of 4.34% (for 1) and 4.20% (for 2) and 4.03% (for 3) in 30–180 °C, is assigned to the removal of one acetone molecule (calculated 3.93% for 1, 3.92% for 2, 3.91% for 3, respectively). The obvious weight loss occurred from 320 to 900 °C for 1–3, accompanying the decomposition of organic ligands, which may decompose into metallic oxides (Fig. S3†). As shown in Fig. S4,† the XRPD experimental patterns of complexes 1–3 are consistent with the simulated ones, confirming the purity of the bulk synthesized materials.
Magnetic properties
Static magnetic properties. The direct current (dc) magnetic behaviors for complexes 1–3 were measured in an applied magnetic field of 1 kOe from 2.0 K to 300 K (Fig. 2). For 1, the χMT value at the room temperature is 9.41 cm3 K mol−1, which is slightly higher than the expected value of 8.88 cm3 K mol−1 for uncoupled NiII (S = 1, g = 2) and GdIII ions (S = 7/2, g = 2). On cooling, χMT product remains nearly constant over the range of 300–50 K. Then, χMT increases sharply to a maximum value of 12.71 cm3 K mol−1 at 2.5 K, close to the value of 12.38 cm3 K mol−1 expected for an isolated S = 9/2 spin, indicating a ferromagnetic coupling between the NiII and GdIII ions of 1. Below this temperature, χMT drops again to a minimum value of 12.61 cm3 K mol−1 at 2.0 K, which may be ascribed to the net spins in a stable state and the spin component along the external field not being changed any longer.20 The experimental data between 300 and 7 K were analyzed on the basis of a simple expression derived from a spin Hamiltonian H = −JSNi·SGd.18,21 The best fit parameters were g = 2.05 and J = 0.80 cm−1 (R = 2.3 × 10−4). The positive J value is in the range ∼0.2–5 cm−1 for phenoxo-bridged NiII–GdIII heterodinuclear complexes, which indicates the ferromagnetic coupling between NiII and GdIII ions.22 The field dependence of the magnetization for 1 was also measured at 1.9 K (Fig. 3), which is qualitatively reproduced by Brillouin curves for S = 9/2,23 further confirming that the spin ground state is derived from the intramolecular ferromagnetic interaction between NiII and GdIII ions. Magnetic entropy change (ΔSm) of 1 was also calculated from the M versus H data to evaluate the magnetocaloric effect (MCE).24 ΔSm can be calculated by using the Maxwell relation ΔSm (T) = ∫[∂M(T,H)/T]HdH from 1.9 K to 5.0 K. The maximum value of −ΔSm is 12.51 J kg−1 K−1 for a field change ΔH = 70 kOe at 1.9 K (Fig. S5†).
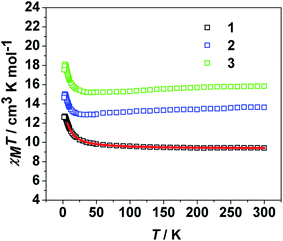 |
| | Fig. 2 Temperature dependence of the χMT values for 1–3 in a 1 kOe field. | |
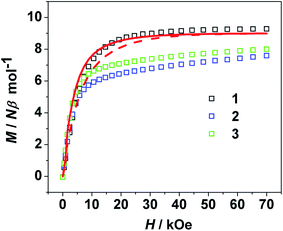 |
| | Fig. 3 Field-dependent magnetizations for 1–3 from 0 to 70 kOe at 1.9 K. The red lines represent the Brillouin function, which correspond to the S = 9/2 (solid line) and non-interacting S = SNi + SGd (dashed line) states with g = 2.05. | |
For 2 and 3, the χMT values at the room temperature are 13.64 and 15.83 cm3 K mol−1, respectively, coincide with the expected values of 12.82 and 15.17 cm3 K mol−1 for uncoupled NiII (S = 1, g = 2) and LnIII (TbIII, J = 6, g = 3/2; DyIII, J = 15/2, g = 4/3) ions (Fig. 2). As lowing temperature, the χMT values decrease gradually to 12.87 cm3 K mol−1 at 35 K for 2, 15.19 cm3 K mol−1 at 40 K for 3. This could be due to the thermal depopulation of the LnIII ions excited Stark sublevels.25 Upon further cooling, χMT increases sharply to a maximum value of 15.01 cm3 K mol−1 at 3.0 K for 2, 18.09 cm3 K mol−1 at 3.0 K for 3, indicating that the strong enough ferromagnetic couplings between NiII and LnIII ions overwhelm the thermal depopulation of LnIII ions. Below this temperature, χMT drops abruptly to a minimum value of 14.65 cm3 K mol−1 for 2, 17.55 cm3 K mol−1 for 3, respectively. Some additional data are also presented as the plots of M versus H in Fig. 3. As the applied magnetic field intensity increases, the magnetization increases rapidly before 10 kOe and then increases slowly to 7.75, 8.00 Nβ at 70 kOe for 2 and 3, without saturation, suggests the presence of magnetic anisotropy and/or the crystal field effects on the LnIII ions.26
Dynamic magnetic properties. Alternating current (ac) magnetic susceptibilities under Hac = 2 Oe were performed for complexes 2 and 3 to check for any SMM behavior. For complex 2, however, no χ′′ peaks were detected even under an external dc field (Fig. S12 and 13†). In the case of 3, the obvious temperature- and frequency-dependent out-of-phase signals (χ′′) were observed as typical SMM behavior. The bistability of the ground state and large intrinsic anisotropy of DyIII ion are crucial to construct SMMs. At zero dc field, the peak maxima of χ′′ were observed below 2.2 K at high frequencies as χ′′ versus v plots depicted in Fig. S6.† The absence of full peaks in χ′′ versus T plots indicates the existence of a fast relaxation process (Fig. S7†), mostly resulting from QTM, which is often revealed in lanthanide SMMs.27 The application of an external dc field can efficiently suppress the strong QTM and an 800 Oe field was found to be optimum for slowing down the relaxation process of 3 (Fig. S8†). When an 800 Oe external field was introduced, the well-shaped peaks of χ′′ were fully observed (Fig. 4). The resulting data were fitted to a Arrhenius law [τ = τ0exp(Δ/kBT)] to afford the anisotropic energy barrier estimated (Δ/kB) of 13.6 K and the pre-exponential factor τ0 = 7.55 × 10−6 s (R = 0.9912) (Fig. 4 inset), confirming a field-induced SMM behavior. The frequency-dependent χ′′ signals exhibit gradual sweep toward the low-frequency region as the temperature was lowered to 1.9 K (Fig. 5) and Cole–Cole plots formed a symmetrically semicircular shape (Fig. 6). The generalized Debye model28 allows to extract the parameter α = 0.167–0.062 for temperatures between 1.9 and 5.0 K (Table S2†), indicating a substantially single relaxation process.29
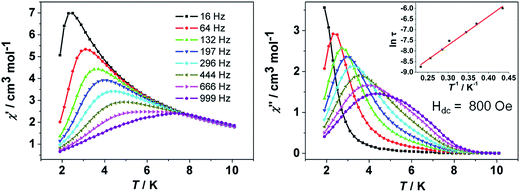 |
| | Fig. 4 Temperature-dependent in-phase χ′ and out-of-phase χ′′ ac susceptibility signals for 3 at the indicated frequencies under 800 Oe dc field. The inset is the Arrhenius fit for the lnτ vs. T−1 plot. | |
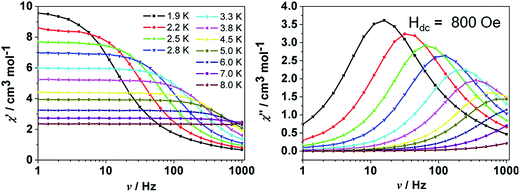 |
| | Fig. 5 Frequency-dependent in-phase χ′ and out-of-phase χ′′ ac susceptibility signals for 3 at the indicated frequencies under 800 Oe dc field. | |
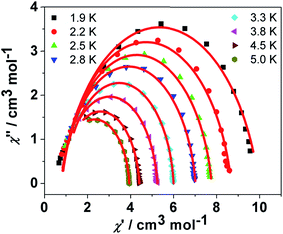 |
| | Fig. 6 Cole–Cole plots for 3 under 800 Oe dc field. The solid lines represent the fit to the Debye model at the indicated temperatures. | |
In order to know the influence of intermolecular dipole–dipole interactions on the magnetic behavior, we also prepared the doped sample (3′) in which complex 3 was diluted by the isostructural YIII analogue in a Dy/Y molar ratio of 1:10 (Fig. S9;† the exact molar ratio of Dy/Y present in the magnetically diluted sample was 0.071:0.929 extracted from ICP measurement). The dc magnetic data of 3′ were measured in detail and the well-shaped peaks of χ′′ were fully observed either in the plots of χ′′ versus T (Fig. 7) or χ′′ versus v (Fig. S10†) under zero dc field. The energy barrier extracted from the data modeled with the Arrhenius law is 18.1 K, with a relaxation time of 7.00 × 10−7 s (R = 0.9965) (Fig. 7 inset), suggesting the doped sample 3′ features SMM behavior under zero dc field. Cole–Cole plots, which show semicircular shapes within the temperature range from 1.9 to 3.5 K, are fitted to a generalized Debye model28 to give values of α in the range of 0.239–0.108 (Fig. S11†). The relatively large α values might indicate the presence of multiple relaxation processes. Interestingly, the adiabatic susceptibility (χS) for the diluted sample (3′) reduces regularly with the increase of temperature (Table S3†),30 which is different from that for 3 (Table S2†). We think it is likely to be due to the diamagnetic dilution that remarkably weakened the dipole–dipole interaction between the molecules.25,31 Further analysis is needed to investigate the relaxation paths in 3′.
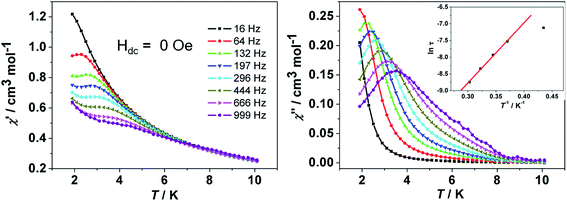 |
| | Fig. 7 Temperature-dependent in-phase χ′ and out-of-phase χ′′ ac susceptibility signals for 3′ at the indicated frequencies under zero dc field. The inset is the Arrhenius fit for the lnτ vs. T−1 plot. | |
It is worth mentioning that the dynamic magnetic behavior of 3 is different from that of one related NiII–DyIII dinuclear complex, [(NiL1)Dy(hfac)2(EtOH)] (Hhfac = hexafluoroacetylacetone).9b [(NiL1)Dy(hfac)2(EtOH)] is also ferromagnetically coupled and shows a slow magnetic relaxation. However, the χ′′ signal is quite weak and SMM behavior cannot be drawn effectively. Meanwhile, complex 3 exhibits strong temperature- and frequency-dependence, a characteristic behavior of SMM. In these two complexes, the octahedral coordination spheres of NiII ions are the same, but DyIII ions adopt two significantly different coordination geometries. Further analyses of the geometries of DyIII ions are determined by SHAPE 2.1 software. For [(NiL1)Dy(hfac)2(EtOH)], DyIII ion is eight-coordinated and lies in a triangular dodecahedron (D2d) symmetry, while DyIII ion is seven-coordinated and the coordination symmetry is close to pentagonal bipyramid (D5h) in 3 (Table S4†). Introduction the thiacalix[4]arene ligand alters the structure and crystal field resulting in significantly different magnetic properties. Further compared to another similar NiII–DyIII complex, [Ni(CH3CN)(H2O)(valpn)Ln(NO3)(H2O)3] (H2 valpn = 1,3-propanediylbis(2-iminomethylene-6-methoxy-phenol)),22 in which the maximum χ′′ value is seen above 2 K only for the highest frequency 1490 Hz under 1000 Oe dc field, we can conclude the thiacalix[4]arene derivatives, which have large steric hindrance and rigid conformation which afford a “protective” sheath and weaken intermolecular f–f interactions,10a are good ligand candidates for the construction of lanthanide-based SMMs.
Conclusions
In summary, three thiacalix[4]arene-supported NiII–LnIII heterodinuclear complexes were successfully synthesized. All the complexes feature diphenoxo-bridged NiII–LnIII dinuclear species. Detailed magnetic studies reveal weak ferromagnetic coupling between NiII and LnIII ions. Complex 3 exhibits a field-induced slow magnetic relaxation with an effective thermal barrier of 13.8 K and a relaxation time of 7.55 × 10−6 s, which is typical for SMMs. Furthermore, the magnetic relaxation behavior of diluted sample with a molar ratio Dy/Y of 1:10, reveals magnetic dilution can efficiently reduce the QTM incurred by intermolecular dipolar interactions. The stepwise synthetic strategy is an efficient approach to construct new 3d–4f heterodinuclear complexes with thiacalix[4]arene and other types of tetradentate ligands (such as porphyrins and phthalocyanines), which is of interest for synthetic chemistry. Additionally, the introduction of thiacalix[4]arene ligand into the heterodinuclear complexes is useful for interesting single-molecule magnets.
Acknowledgements
This work was supported by the Major State Basic Research Development Program (2013CB922101), the National Natural Science Foundation of China (51173075 and 91433113), and the Natural Science Foundation of Jiangsu Province (BK20130054). We also thank Dr Tian-Wei Wang for the experimental assistance in conducting magnetic measurements.
References
-
(a) F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119 RSC;
(b) R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS;
(c) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS;
(d) K. Binnemans, Coord. Chem. Rev., 2009, 109, 4283 CrossRef CAS PubMed;
(e) K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396 RSC;
(f) T. D. Harris, M. V. Bennett, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2010, 132, 3980 CrossRef CAS PubMed;
(g) K. Qian, X. C. Huang, C. Zhou, X. Z. You, X. Y. Wang and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 13302 CrossRef CAS PubMed;
(h) M. Morimoto, H. Miyasaka, M. Yamashita and M. Irie, J. Am. Chem. Soc., 2009, 131, 9823 CrossRef CAS PubMed.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420 CrossRef CAS PubMed.
-
(a) L. Chen, J. Wang, J. M. Wei, W. Wernsdorfer, X. T. Chen, Y. Q. Zhang, Y. Song and Z. L. Xue, J. Am. Chem. Soc., 2014, 136, 12213 CrossRef CAS PubMed;
(b) Y. N. Guo, G. F. Xu, P. Gamez, L. Zhao, S. Y. Lin, R. Deng, J.-K. Tang and H. J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538 CrossRef CAS PubMed;
(c) J. Long, R. Vallat, R. A. Ferreira, L. D. Carlos, F. A. Almeida Paz, Y. Guari and J. Larionova, Chem. Commun., 2012, 48, 9974 RSC;
(d) J. Rinck, G. Novitchi, W. van den Heuvel, L. Ungur, Y. Lan, W. Wernsdorfer, C. E. Anson, L. F. Chibotaru and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 7583 CrossRef CAS PubMed;
(e) L. Rosado Piquer and E. C. Sañudo, Dalton Trans., 2015, 44, 8771 RSC.
-
(a) F. H. Zhao, H. Li, Y. X. Che, J. M. Zheng, V. Vieru, L. F. Chibotaru, F. Grandjean and G. J. Long, Inorg. Chem., 2014, 53, 9785 CrossRef CAS PubMed;
(b) G. Rogez, J. N. Rebilly, A. L. Barra, L. Sorace, G. Blondin, N. Kirchner, M. Duran, J. van Slageren, S. Parsons, L. Ricard, A. Marvilliers and T. Mallah, Angew. Chem., Int. Ed., 2005, 44, 1876 CrossRef CAS PubMed.
-
(a) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed;
(b) Y. N. Guo, G. F. Xu, Y. Guo and J.-K. Tang, Dalton Trans., 2011, 40, 9953 RSC;
(c) P.-H. Guo, J. Liu, Z.-H. Wu, H. Yan, Y.-C. Chen, J.-H. Jia and M.-L. Tong, Inorg. Chem., 2015, 54, 8087 CrossRef CAS PubMed.
-
(a) E. Colacio, J. Ruiz-Sanchez, F. J. White and E. K. Brechin, Inorg. Chem., 2011, 50, 7268 CrossRef CAS PubMed;
(b) J.-F. Wu, L. Zhao, P. Zhang, L. Zhang, M. Guo and J.-K. Tang, Dalton Trans., 2015, 44, 11935 RSC;
(c) X. L. Li, F. Y. Min, C. Wang, S. Y. Lin, Z. Liu and J.-K. Tang, Inorg. Chem., 2015, 54, 4337 CrossRef CAS PubMed;
(d) M. Towatari, K. Nishi, T. Fujinami, N. Matsumoto, Y. Sunatsuki, M. Kojima, N. Mochida, T. Ishida, N. Re and J. Mrozinski, Inorg. Chem., 2013, 52, 6160 CrossRef CAS PubMed;
(e) J.-L. Liu, J.-Y. Wu, Y.-C. Chen, V. Mereacre, A. K. Powell, L. Ungur, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2014, 53, 12966 CrossRef CAS PubMed.
-
(a) W.-B. Sun, B. Yan, Y.-Q. Zhang, B.-W. Wang, Z.-M. Wang, J.-H. Jia and S. Gao, Inorg. Chem. Front., 2014, 1, 503 RSC;
(b) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092 RSC;
(c) J. Liu, Y.-C. Chen, Z.-X. Jiang, J.-L. Liu, J.-H. Jia, L.-F. Wang, Q.-W. Li and M.-L. Tong, Dalton Trans., 2015, 44, 8150 RSC.
-
(a) X. Feng, W. Zhou, Y. Li, H. Ke, J.-K. Tang, R. Clérac, Y. Wang, Z. Su and E. Wang, Inorg. Chem., 2012, 51, 2722 CrossRef CAS PubMed;
(b) P. Bag, J. Goura, V. Mereacre, G. Novitchi, A. K. Powellb and V. Chandrasekhar, Dalton Trans., 2014, 43, 16366 RSC;
(c) M. Andruh, J.-P. Costes, C. Diaz and S. Gao, Inorg. Chem., 2009, 48, 3342 CrossRef CAS PubMed;
(d) H. L. C. Feltham, R. Clérac, A. K. Powell and S. Brooker, Inorg. Chem., 2011, 50, 4232 CrossRef CAS PubMed.
-
(a) T. Yamaguchi, Y. Sunatsuki and H. Ishida, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, m156 CAS;
(b) T. Yamaguchi, Y. Sunatsuki, H. Ishida, M. Kojima, H. Akashi, N. Re, N. Matsumoto, A. Pochaba and J. Mroziński, Inorg. Chem., 2008, 47, 5736 CrossRef CAS PubMed;
(c) T. Yamaguchi, J.-P. Costes, Y. Kishima, M. Kojima, Y. Sunatsuki, N. Bréfuel, J.-P. Tuchagues, L. Vendier and W. Wernsdorfer, Inorg. Chem., 2010, 49, 9125 CrossRef CAS PubMed.
-
(a) F. Gao, L. Cui, Y. Song, Y.-Z. Li and J.-L. Zuo, Inorg. Chem., 2014, 53, 562 CrossRef CAS PubMed;
(b) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824 RSC;
(c) Y.-F. Bi, X.-T. Wang, W.-P. Liao, X.-W. Wang, R.-P. Deng, H.-J. Zhang and S. Gao, Inorg. Chem., 2009, 48, 11743 CrossRef CAS PubMed;
(d) R. E. Fairbairn, R. McLellan, R. D. McIntosh, M. A. Palacios, E. K. Brechin and S. J. Dalgarno, Dalton Trans., 2014, 43, 5292 RSC;
(e) S. Kennedy, C. M. Beavers, S. J. Teat and S. J. Dalgarno, CrystEngComm, 2014, 16, 3712 RSC;
(f) P. K. Thallapally, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2006, 128, 15060 CrossRef CAS PubMed.
-
(a) K. Su, F. Jiang, J. Qian, L. Chen, J. Pang, S. M. Bawaked, M. Mokhtar, S. A. Al-Thabaiti and M.-C. Hong, Inorg. Chem., 2015, 54, 3183 CrossRef CAS PubMed;
(b) K. Su, F. Jiang, J. Qian, M. Wu, K. Xiong, Y. Gai and M.-C. Hong, Inorg. Chem., 2013, 52, 3780 CrossRef CAS PubMed;
(c) J.-Y. Ge, J. Ru, F. Gao, Y. Song, X.-H. Zhou and J.-L. Zuo, Dalton Trans., 2015, 44, 15481 RSC.
- S. Ghosh, Y. Ida, T. Ishida and A. Ghosh, Cryst. Growth Des., 2014, 14, 2588 CAS.
- H. Dvorakova, J. Lang, J. Vlach, J. Sykora, M. Cajan, M. Himl, M. Pojarova, I. Stibor and P. Lhotak, J. Org. Chem., 2007, 72, 7157 CrossRef CAS PubMed.
- E. A. Boudreaux and L. N. Mulay, Theory and Application of Molecular Paramagnetism, John Wiley & Sons, New York, 1976, p. 491 Search PubMed.
- SAINT-Plus, version 6.02, Bruker Analytical X-ray System, Madison, WI, 1999 Search PubMed.
- G. M. Sheldrick, SADABS an empirical absorption correction program, Bruker Analytical X-ray Systems, Madison, WI, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- X.-Y. Lu, Y.-Q. Liu, X.-W. Deng, Z.-X. Zhu, M.-X. Yao and S. Jing, New J. Chem., 2015, 39, 3467 RSC.
- T. D. Pasatoiu, M. Etienne, A. M. Madalan, M. Andruh and R. Sessoli, Dalton Trans., 2010, 39, 4802 RSC.
- J.-H. Wang, P.-F. Yan, G.-M. Li, J.-W. Zhang, P. Chen, M. Suda and Y. Einaga, Inorg. Chim. Acta, 2010, 363, 3706 CrossRef CAS.
- J. P. Costes, F. Dahan, A. Dupuis and J. P. Laurent, Inorg. Chem., 1997, 36, 4284 CrossRef CAS.
- T. D. Pasatoiu, J. P. Sutter, A. M. Madalan, F. Z. Fellah, C. Duhayon and M. Andruh, Inorg. Chem., 2011, 50, 5890 CrossRef CAS PubMed.
- M. G. Alexandru, D. Visinescu, S. Shova, F. Lloret, M. Julve and M. Andruh, Inorg. Chem., 2013, 52, 11627 CrossRef CAS PubMed.
-
(a) M. Manoli, A. Collins, S. Parsons, A. Candini, M. Evangelisti and E. K. Brechin, J. Am. Chem. Soc., 2008, 130, 11129 CrossRef CAS PubMed;
(b) J. Ruiz, G. Lorusso, M. Evangelisti, E. K. Brechin, S. J. A. Pope and E. Colacio, Inorg. Chem., 2014, 53, 3586 CrossRef CAS PubMed;
(c) J. W. Sharples, Y.-Z. Zheng, F. Tuna, E. J. L. McInnes and D. Collison, Chem. Commun., 2011, 47, 7650 RSC;
(d) W. Sethi, S. Sanz, K. S. Pedersen, M. A. Sørensen, G. S. Nichol, G. Lorusso, M. Evangelisti, E. K. Brechin and S. Piligkos, Dalton Trans., 2015, 44, 10315 RSC;
(e) M. Manoli, R. D. L. Johnstone, S. Parsons, M. Murrie, M. Affronte, M. Evangelisti and E. K. Brechin, Angew. Chem., Int. Ed., 2007, 46, 4456 CrossRef CAS PubMed.
- Y. Bi, Y.-N. Guo, L. Zhao, Y. Guo, S.-Y. Lin, S.-D. Jiang, J.-K. Tang, B.-W. Wang and S. Gao, Chem.–Eur. J., 2011, 17, 12476 CrossRef CAS PubMed.
- J. Goura, R. Guillaume, E. Riviere and V. Chandrasekhar, Inorg. Chem., 2014, 53, 7815 CrossRef CAS PubMed.
-
(a) G. Brunet, F. Habib, I. Korobkov and M. Murugesu, Inorg. Chem., 2015, 54, 6195 CrossRef CAS PubMed;
(b) E. Moreno Pineda, N. F. Chilton, F. Tuna, R. E. P. Winpenny and E. J. L. McInnes, Inorg. Chem., 2015, 54, 5930 CrossRef CAS PubMed.
- K. Katoh, T. Kajiwara, M. Nakano, Y. Nakazawa, W. Wernsdorfer, N. Ishikawa, B. K. Breedlove and M. Yamashita, Chem.–Eur. J., 2011, 17, 117 CrossRef CAS PubMed.
- M.-X. Yao, Q. Zheng, K. Qian, Y. Song, S. Gao and J.-L. Zuo, Chem.–Eur. J., 2013, 19, 294 CrossRef CAS PubMed.
-
(a) V. Chandrasekhar, S. Hossain, S. Das, S. Biswas and J.-P. Sutter, Inorg. Chem., 2013, 52, 6346 CrossRef CAS PubMed;
(b) R. J. Blagg, F. Tuna, E. J. L. McInnes and R. E. P. Winpenny, Chem. Commun., 2011, 47, 10587 RSC.
-
(a) S. Titos-Padilla, J. Ruiz, J. M. Herrera, E. K. Brechin, W. Wersndorfer, F. Lloret and E. Colacio, Inorg. Chem., 2013, 52, 9620 CrossRef CAS PubMed;
(b) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. McInnes, L. F. Chibotaru and R. E. Winpenny, Nat. Chem., 2013, 5, 673 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, TGA curves, XRPD patterns, magnetic characterizations and X-ray crystallographic files in CIF format for 1–3. CCDC 1424163–1424165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra23697e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 







