
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect delamination of graphite ore into defect-free graphene using a biphasic solvent system under pressurized ultrasound†
H.
Beneš
a,
R. K.
Donato
b,
P.
Ecorchard
*c,
D.
Popelková
ac,
E.
Pavlová
a,
D.
Schelonka
c,
O.
Pop-Georgievski
a,
H. S.
Schrekker
*b and
V.
Štengl
c
aInstitute of Macromolecular Chemistry AS CR, v.v.i, Heyrovsky Sq. 2, 16206, Prague 6, Czech Republic
bLaboratory of Technological Processes and Catalysis, Institute of Chemistry, Universidade Federal do Rio Grande do Sul-UFRGS, Av. Bento Gonçalves 9500, Porto Alegre, RS, Brazil. E-mail: henri.schrekker@ufrgs.br
cInstitute of Inorganic Chemistry AS CR, v.v.i, 25068 Řež, Czech Republic. E-mail: ecorchard@iic.cas.cz
First published on 8th January 2016
Abstract
The study reported herein describes for the first time a method for producing defect-free graphene directly from its mineral ore. This was achieved by treating graphite ore in an ionic liquid containing biphasic solvent system, applying pressurized intense cavitation ultrasound. Without any graphite ore pre-treatment, large few layer thick graphene sheets, as well as micrometric layered crumpled graphene structures, were produced. This opens new opportunities for the development of graphene-based technologies.
Introduction
Since the 1940s, graphene sheets of interconnected sp2-hybridized carbon atoms have been known.1 Although this material was considered as “unrealistic” due to its synthetic hurdles and low thermodynamic stability at ambient conditions, Geim and Novoselov changed this perspective in 2004.2–4 From this point of showing that graphene is a “possible” material with a huge potential for the development of high-tech applications, intensive research efforts have been focused on enhancing its accessibility.Nowadays, few-layers thick graphene nanosheets can be produced by several methods; such as mechanical exfoliation (repeated peeling),4 solvation force-induced exfoliation,5 and chemical oxidation/reduction6–8 of graphite, unrolling of multiwall carbon nanotubes (MWCNT),9–11 epitaxial growth,12 chemical vapor deposition (CVD),13 electrochemical reaction14 and gas-phase synthesis.15 Common drawbacks of these techniques are very low yields, high-energy demands and/or necessity to use expensive high-quality substrates. Another important aspect to consider is the quality of the produced graphenes. For instance, the chemical reduction of oxidized graphite is incomplete and produces graphenes with residual functional groups, causing the loss of the inherent graphene properties (i.e. electrical, mechanical and thermal properties).16,17 Therefore, the choice of the proper chemicals and processing conditions is essential for obtaining defect-free graphene nanosheets.
Within this context, sonication of graphite has shown to be a powerful tool for the preparation of graphene, although this requires the adjustment of the medium's surface energy close to that of graphene (∼68 mJ m−2).26,27 Besides the medium's surface energy, the strength of the medium-graphene physical interaction is another important parameter. A variety of solvents and solvent combinations that prevent reverse graphene aggregation (e.g. ethylene glycol, dimethylsulfoxide [DMSO], dimethylformamide [DMF], N-methyl-pyrrolidone [NMP], c-butyrolactone, dimethylacetamide, or its mixtures with water) have been successfully applied in the exfoliation of graphite under high-intensity ultrasound waves from an ultrasound horn, producing solvated graphene flakes.18,19 This strategy allows the preparation of graphene monolayer dispersions in DMF,20,21 benzene22 and NMP.19,23 In addition, the medium's surface energy can be adjusted by the addition of a surfactant, which has been achieved with sodium dodecylbenzene sulphonate in a water solution, providing stable graphene suspensions.24 Despite these advances, the maximum achieved graphene sheets concentrations remain low, around 1 mg mL−1.25
Considering the potential use of surfactants and the importance of the physical medium-graphene interaction, imidazolium ionic liquids (IL) open new opportunities. The π-electron cloud of the imidazolium cation provides relatively strong interactions with various π-electronic carbon nanomaterials, including MWCNT, fullerenes and graphene.28 Besides the successful application of imidazolium IL in the unrolling and dispersion of MWCNT,10 a few studies have been reported about its use as a medium in the direct exfoliation of graphite into graphene sheets. Wang et al. employed ultrasound to directly exfoliate graphite flakes in the IL 1-n-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (BMImNTf2), which was followed by high-speed centrifugation to remove the fast sedimenting non-exfoliated graphite.29 The supernatant IL suspension contained up to 0.95 mg mL−1 of graphene sheets. Nuvoli et al. showed that a more concentrated graphene–IL suspension (5.33 mg mL−1) could be obtained when grinding graphite flakes and 1-n-hexyl-3-methylimidazolium hexafluorophosphate in a mortar, before sonication.30 The produced graphene sheets exhibited an average thickness of ∼2 nm (6–7 layers). When using the IL 1-n-butyl-3-methylimidazolium hexafluorophosphate, Shang et al. exfoliated natural graphite by sequential mechanical grinding and sonication into graphene sheets of 2–5 layers.31 Another approach was used by Matsumoto et al., applying a polymeric IL with the hexafluorophosphate anion.32 Microwave irradiation decomposed this anion under the formation of HF molecules, which intercalated within the graphite galleries and promoted exfoliation.
Although significant progress has been achieved in the preparation of graphene, a simple and high-yielding procedure for the large-scale production of cheap defect-free single- or few-layer graphene with large area and smooth edges remains a challenge.26 As this seems being the main requisite to explore the full potential of graphene in the development of high-tech applications,33 this study addresses the development of a simple one-step procedure for the preparation of graphene from the inexpensive untreated graphite ore. For the first time, graphite ore has been completely delaminated, treating an IL containing biphasic solvent system with cavitation in a pressurized ultrasonic reactor. This allowed not only obtaining defect-free graphene, but also enabled the facile segregation of the graphene from the mineral impurities.
Experimental
Materials
A crystalline graphite ore (carbon content of 48 wt%; mined in Lazec – situated in the Southbohemian Moldanubicum region) and a natural graphite powder (carbon content of 96 wt% and flake diameter in the range of 3–30 μm) were kindly provided by Koh-i-noor Grafite, Ltd., Czech Republic. 1-n-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (BMImNTf2; purity ≥ 98%) and 1-octanol (C8H17OH; purity ≥ 99.5%) were purchased from Sigma-Aldrich. Dichloromethane (CH2Cl2; purity ≥ 99.5%) was supplied by Lach-Ner.Graphite exfoliation in high-pressure ultrasound reactor (HP-US)
Graphite ore (0.3 g), together with the biphasic water (95 mL) and organic solvent (CH2Cl2 or C8H17OH, 5 mL) system, and IL BMImNTf2 in the indicated quantity, was added to an ultrasound reactor and pressurized to 5 bar (UIP 2000hd, 20 kHz, 2000 W, Hielscher Ultrasonics GmbH). The reaction vessel was connected to a refrigerated recirculator, keeping the reaction temperature at ∼10 °C. Three different sonication programs were applied, using in all cases 1.3 kW of sonication power: (i) 10 min of uninterrupted exposition, (ii) two 15 min expositions intercalated with a 5 min interval (total sonication time: 30 min), and (iii) two 15 min and one 20 min expositions intercalated with two 5 min intervals (total sonication time: 50 min) (Fig. 1). Following the sonication process, the obtained suspensions were stored in glass vessels. | ||
| Fig. 1 Photographic images of the graphite ore fragment used and the dispersions obtained before pressurized sonication in H2O–CH2Cl2 (a) and after pressurized sonication in H2O–CH2Cl2 (b) and in H2O–C8H17OH (c) systems. Images (b) and (c) were taken 3 months after the vials were stored. | ||
Atomic force microscopy (AFM)
A Dimension Icon Atomic Force Microscope (Bruker) was used to record the AFM images. A silicon tip on a nitride lever was employed for the measurement in ScanAsyst-air contact mode. Two methods were used to deposit the sample on a suitable support prior the measurement. (i) The majority of the samples were prepared by a spin-coating method; where the samples were pipetted (20–40 μL) onto a smooth mica support and spread over the substrate by rotation. (ii) The rest of the samples were prepared by depositing a drop on a glass microscope slide.Transmission electron microscopy (TEM)
TEM samples were deposited onto copper TEM grids (300 mesh) coated with a holey carbon, washed few times with CH2Cl2 and imaged using a Tecnai G2 Spirit Twin at 120 kV (FEI).High resolution transmission electron microscopy (HRTEM)
Samples for HRTEM were first diluted in CH2Cl2 and put into bath ultrasound for 5 min. Then, the diluted sample was deposited onto carbon-coated lacey TEM grid. The solvent was left to evaporate and the sample was dried under dynamic vacuum at 80 °C for 4 hours. HRTEM and selected area electron diffraction (SAED) were carried out using a FEI Talos transmission electron microscope operating at 120 kV.X-ray diffraction (XRD)
Diffraction patterns of thin films on a silicon sample holder were measured using a diffractometer Bruker D2 equipped with a conventional X-ray tube (CuKα radiation, 30 kV, 10 mA). The following conditions were used: 0.6 mm primary divergence slit module width, Soler Module 2.5, 2 mm Airscatter screen module, 0.5 mm Ni Kbeta-filter, 5–90° range, 0.00405° steps; 0.3 s per step, and the LYNXEYE 1-dimensional detector. The crystallite size, interlayer spacing and number of C atom layers were calculated using the Debye–Scherrer equations.34,35Raman spectroscopy
The Raman spectra were taken on a DXR Raman microscope (Thermo Scientific) using a 10× objective lens (Olympus microscope) with a 532 nm (3 mW) laser excitation, being the average of 32 two-second scans. Graphene/graphite dispersions were dropped on plasma-treated microscopic glasses, 3 times washed with CH2Cl2 and dried at 60 °C.X-ray photoelectron spectroscopy (XPS)
Thin films were prepared from CH2Cl2-diluted graphene/graphite dispersions on plasma-treated microscopic glasses, followed by drying under vacuum at 80 °C. The XPS measurements were carried out on a K-Alpha+ XPS spectrometer (ThermoFisher Scientific) operating at a base pressure of 1.0 × 10−7 Pa. The data acquisition and processing were performed using the Thermo Avantage software. All samples were analyzed using a microfocused, monochromated Al Kα X-ray radiation (400 μm spot size) with a pass energy of 200 eV for survey and 50 eV for high-energy resolution core level spectra. The X-ray angle of incidence was 30° and the emission angle was along the surface normal. The K-Alpha charge dual compensation system was employed during analysis, using electrons and low-energy argon ions to prevent any localized charge build-up. The obtained high-resolution spectra were fitted/deconvoluted with Voigt profiles (binding energy uncertainty: ±0.1 eV). The analyzer transmission function, Scofield sensitivity factors, and effective attenuation lengths (EAL) for photoelectrons were applied for quantification. EAL were calculated using the standard TPP-2M formalism. All spectra were referenced to the C 1s peak attributed to C–C, C–H at 285.0 eV binding energy, which were controlled by means of the well-known photoelectron peaks of metallic Cu, Ag, and Au.Results and discussion
Biphasic solvent systems, composed of organic (CH2Cl2 or C8H17OH) and water phase, were used as medium for graphite ore delamination to graphene sheets in a high-pressure ultrasound reactor (HP-US). At the same time, the graphite flakes were isolated from the country rock due to the differences in surface tension. Contrary to the conventional flotation techniques where only the mineral impurities attached to the surface of graphite flakes are removed,36 in this system the “intercalated” ash that resides between adjacent graphene layers' stack is separated from the formed graphene sheets. As demonstrated in Fig. 1, the purified graphene sheets were mainly trapped on water–organic solvent interphase, covered the organic solvent–glass interface and partially located in the organic phase, whilst the mineral impurities remained in the water phase. The use of organic solvents with different densities allowed choosing where the phase with obtained graphene was i.e., CH2Cl2 is denser than water and graphene stayed from the interphase to the lower phase (Fig. 1b), while C8H17OH is less dense than water trapping graphene from interphase to the upper phase (Fig. 1c).The influence of the particles wettability by the liquids is a first order parameter in the particles adsorption behavior at the fluid–fluid interphase. When omitting any kinetic effects, the preferential adsorption of the formed graphene particles on the water–organic solvent interphase, in the equilibrium state, is governed by thermodynamics' tendency to minimize the interfacial energy.37 The knowledge of interfacial tensions between both liquids and between liquids and graphene should be sufficient to calculate wettability parameter and to predict the graphene's location in the biphasic system. The calculated wettability parameter predicts the graphene location at the interphase, which was confirmed by the experiments (the details are given in the ESI†). Preferentially, high aspect ratio planar graphene sheets adsorb at the liquid–liquid interphase, which has also been described in the literature for water–chloroform38 and water–heptane39 systems. Interestingly, no sedimentation and re-aggregation could be observed in the dispersions after storage for 3 months (Fig. 1), indicating that the interfacial graphene layers were very stable and energetically more favorable than restacking and agglomeration of the graphene sheets.38
Imidazolium IL have demonstrated huge potential as medium in the unrolling of MWCNT and the exfoliation of graphite.10 Especially, the hydrophobic IL BMImNTf2 appears being extremely compatible with these approaches, acting not only as a surfactant for carbon nanobjects through π–π stacking (graphene–imidazolium cation),40 but also as the “unrolling/exfoliating” agent when applying specific sonication conditions.10 In the current study, a similar effect of BMImNTf2 was observed on the graphite ore exfoliation and dispersion when applied as additive. The delamination kinetics in the absence and presence of 0.3 wt% of BMImNTf2 were investigated using TEM and XRD techniques (Fig. 2).
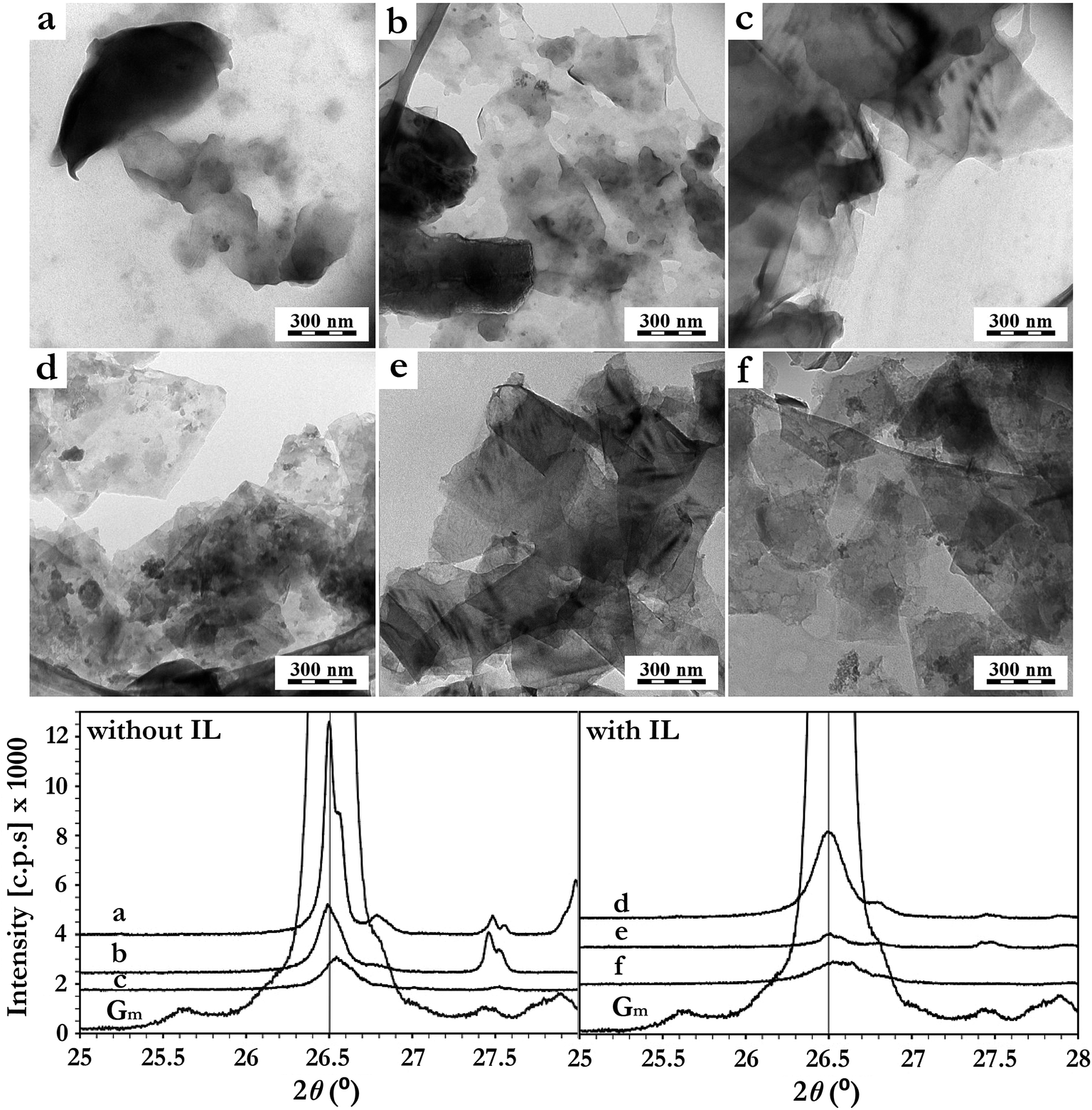 | ||
| Fig. 2 TEM images and XRD diffractograms of dispersed graphite–graphene after 10 min (a), 30 min (b) and 50 min (c) sonication in the H2O–CH2Cl2 system, and after 10 min (d), 30 min (e) and 50 min (f) sonication in the H2O–CH2Cl2–IL system. The XRD pattern of pristine graphite ore (Gm) is given for comparison. | ||
According to the TEM images, no graphite ore delamination was observed after 10 min of sonication in the absence of IL (Fig. 2a). For this case, the total sonication time had to be prolonged up to 50 min (Fig. 2c) to produce graphene sheets. This exfoliation progress was also observed by XRD (Fig. 2). The graphite diffraction pattern contains a typical (002) reflection at 2θ = 26.4°, corresponding to the basal distance of 0.336 nm. Its diffraction intensity (002) decreased progressively after 30 min and 50 min of pressurized sonication (Fig. 2). On the other hand, the IL containing system showed a much more efficient graphite delamination, as graphene sheets were already formed after 10 min of sonication (Fig. 2d). The XRD diffractogram is in agreement with this observation, presenting already a drastic decrease in the intensity of the (002) diffraction peak after 10 min (Fig. 2). After 30 min, the (002) peak intensity was much lower compared to those measured from samples without IL, confirming the beneficial role of BMImNTf2 in this process.
In spite of the samples dilution and washing with CH2Cl2 prior to TEM analyses, the presence of IL (aggregated gray spots) on the graphene sheets was observed. This is in agreement with the strong π–π interaction between the imidazolium ring of BMImNTf2 and the graphene surface.10,29,30,41 The physical adsorption of IL on the graphene sheets surface was further investigated using XPS (Table 1). An increase in the nitrogen content and the appearance of sulfur and fluorine indicated the BMImNTf2 presence on the graphene surface. Notably, the N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F:S ratio (considering the nitrogen increment due to the presence of IL) was 1.5:3.0:1.1 which was in excellent agreement with the expected theoretical ratio for BMImNTf2 (1.5:3.0:1.0). Moreover, the heteroatom–carbon proportion obtained corresponds to that of the IL's molecular composition. Considering that BMImNTf2 has the molecular formula C10H15F6N3O4S2, the expected C1s:O1s ratio of neat IL is 2.5. The obtained C1s:O1s for exfoliated graphite without and with IL were 4.57 and 2.64, respectively. As a consequence, it is reasonable to infer that most of the atomic contribution obtained by XPS was from the IL, thus explaining the increased O1s values and excluding the possibility of an oxidative process. The high-resolution C1s, N1s, S2p and F1s spectra of exfoliated graphite ore can be found in the ESI (Fig. S1†).
:F:S ratio (considering the nitrogen increment due to the presence of IL) was 1.5:3.0:1.1 which was in excellent agreement with the expected theoretical ratio for BMImNTf2 (1.5:3.0:1.0). Moreover, the heteroatom–carbon proportion obtained corresponds to that of the IL's molecular composition. Considering that BMImNTf2 has the molecular formula C10H15F6N3O4S2, the expected C1s:O1s ratio of neat IL is 2.5. The obtained C1s:O1s for exfoliated graphite without and with IL were 4.57 and 2.64, respectively. As a consequence, it is reasonable to infer that most of the atomic contribution obtained by XPS was from the IL, thus explaining the increased O1s values and excluding the possibility of an oxidative process. The high-resolution C1s, N1s, S2p and F1s spectra of exfoliated graphite ore can be found in the ESI (Fig. S1†).
| Orbital | Exfoliated graphite ore | Exfoliated graphite ore + IL | ||
|---|---|---|---|---|
| Atomic% | Weight% | Atomic% | Weight% | |
| S2p | 0.0 | 0.0 | 1.1 ± 0.3 | 6.1 ± 0.6 |
| C1s | 81.4 ± 0.3 | 76.7 ± 0.4 | 69.6 ± 1.4 | 63.3 ± 1.4 |
| N1s | 0.8 ± 0.1 | 0.9 ± 0.1 | 2.3 ± 0.2 | 2.7 ± 0.2 |
| O1s | 17.8 ± 0.3 | 22.4 ± 0.3 | 26.4 ± 2.6 | 25.3 ± 3.0 |
| F1s | 0.0 | 0.0 | 3.0 ± 0.6 | 5.2 ± 0.9 |
The degree of graphite exfoliation was determined by AFM (Fig. 3). Graphene flakes with an average thickness of 1–1.5 nm were obtained after pressurized sonication for 50 min without IL addition (Fig. 3c and f). When IL was added, graphene flakes with a thickness of <1.5 nm were produced within a much shorter sonication time of 10 min (Fig. 3a and d). Additional sonication (30 min in total) led to further thickness reduction of the graphene flakes to ∼1.0 nm (Fig. 3b and e). Considering the thickness of single graphene layer (0.335 nm),42 10 min and 30 min of pressurized sonication in the presence of IL led to graphene flakes with less than 5 and 3 layers, respectively. As the XPS analyses confirmed the presence of IL on the graphene surfaces (Table 1), their presence on the graphene sheets, as well as between the mica substrate and graphene sheets, cannot be excluded. Thus, the amount of graphene layers could in fact be smaller, indicating the formation of bilayers and monolayers. Furthermore, Hernandez et al. observed on AFM images thicker graphene monolayers than expected (1–2 nm instead of 0.3 nm).19 Generally, the thicknesses ≤ 1 nm determined from AFM height profiles are consistent with the formation of graphene.43–45
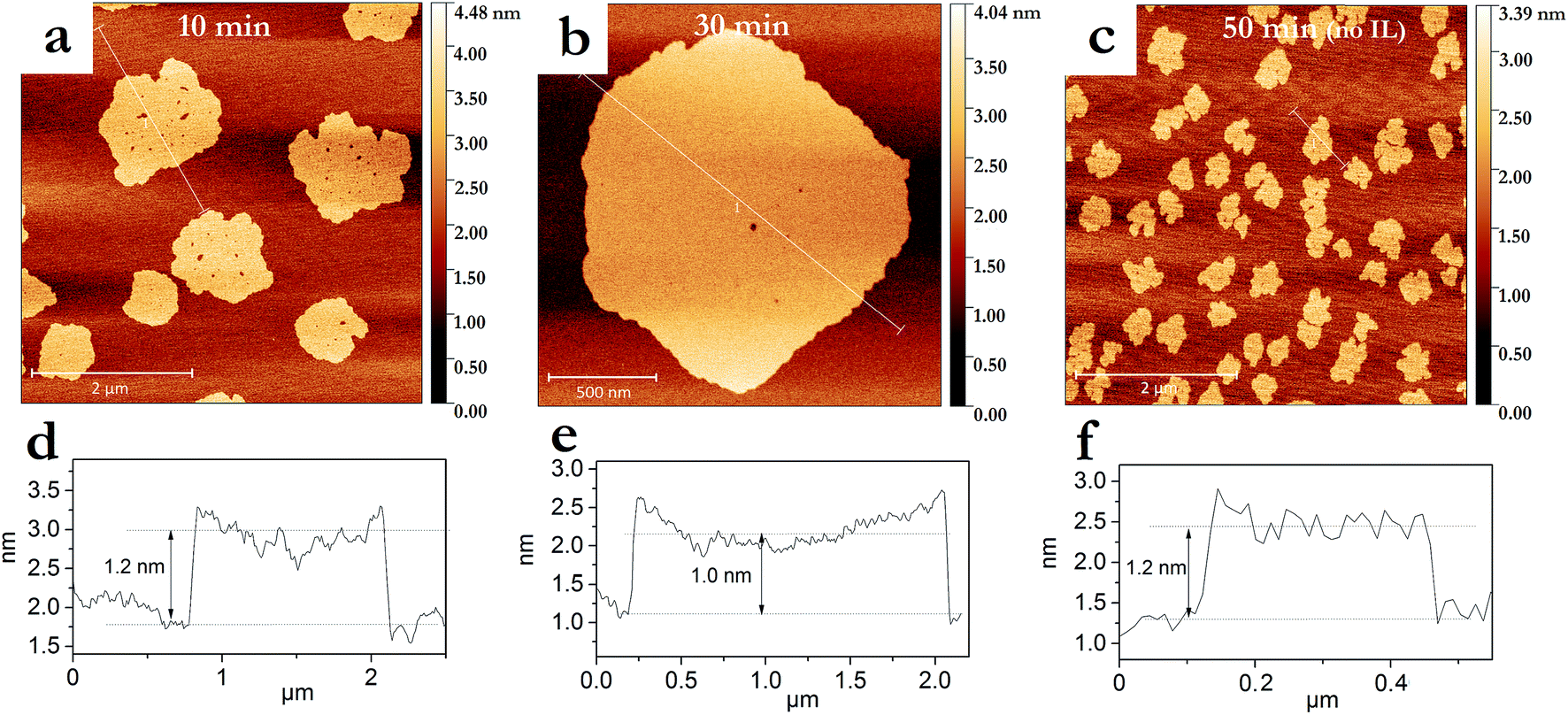 | ||
| Fig. 3 AFM topology images of graphene sheets obtained after 10 min (a) and 30 min (b) pressurized sonication in the H2O–CH2Cl2–IL system, and (c) 50 min of pressurized sonication in the H2O–CH2Cl2 system. The graphs (d)–(f) are the height profiles along the lines displayed in (a)–(c), respectively. | ||
The AFM study provided further information about the dimensions of the obtained graphenes. IL-assisted pressurized sonication produced after 10 min graphene sheets with much larger lateral dimensions (average diameter of 1100 ± 350 nm) compared to those obtained after 50 min in an IL-free system (average diameter of 380 ± 75 nm) (Fig. 3). This reduction in the lateral dimensions could indicate transversal breakage due to a too long exposition to intense cavitation.46 The IL seems to help dissipating the excessive cavitation energy, since a similar phenomenon of graphene sheets lateral size reduction was only observed after 50 min of exposition, and in a much less intense manner (Fig. 2f).
The quality of the graphene sheets produced by the best evaluated procedure (30 min sonication in the presence of IL) was further evaluated by TEM (Fig. 4). Confirming the previous characterizations, mostly few-layers (Fig. 4a) and monolayer (Fig. 4b) graphene were formed. HRTEM (Fig. 4c) and two-dimensional fast Fourier transform (FFT) (Fig. 4d) images of the formed layers showed sets of district spots at 0.21 and 0.12 nm, which correspond to periodic hexagonal structure of AB Bernal stacked few-layer graphene.47 The selected-area electron diffraction pattern taken from Fig. 4b is displayed in Fig. 4e, and the diffracted intensity taken along the (1−210)–(0−110)–(−1010)–(−2110) axis for this pattern is displayed in Fig. 4f. The inner peaks, (0−110) and (−1010), were more intense than the outer ones, (1−210) and (−2110), confirming the presence of a graphene monolayer in Fig. 4b.19,48 Besides, all observed areas seemed to be free of structural defects, evidencing that this exfoliation procedure was nondestructive. In the case of the systems with IL, the sheet edges also tended to bend (Fig. 3b) and crumple (Fig. 4a). Moreover, HRTEM images showed regions where fringes were observed, which indicated local curvature of the sheets.49
 | ||
| Fig. 4 Electron microscopy of graphene sheets obtained after 30 min HP-US in H2O–CH2Cl2 in the presence of IL. TEM micrographs of (a) few-layers and (b) monolayer graphene; HRTEM micrograph (c) of a few-layers graphene taken from (a); two-dimensional FFT (d) of few-layers graphene taken from (a); electron diffraction patterns (e) of monolayer graphene taken from (b); diffracted intensity (f) taken along the (1−210)–(0−110)–(−1010)–(−2110) axis from the pattern shown in (e). | ||
The exfoliating experiments were also successfully performed when the H2O–C8H17OH biphasic system was used. As in the case of H2O–CH2Cl2, BMImNTf2 accelerated the whole exfoliation process. The intensity of the XRD 002 peak (2θ = 26.5) decreased to less than 1% of its initial value after 30 minutes of pressurized sonication, indicating structural disordering and graphite exfoliation,32 in a slightly lower degree when compared to the H2O–CH2Cl2 system (Fig. S2†). HRTEM images confirmed the formation of few-layers thick graphene sheets (Fig. S3†).
Raman spectroscopy is another important tool to determine the graphene quality. Fig. S4† shows the Raman spectra of bulk graphite ore and cast films from the graphene suspensions (without and with BMImNTf2). Graphitic materials typically involve three bands; (i) the D-band (∼1350 cm−1), (ii) the G-band (∼1580 cm−1) and (iii) the 2D-band (∼2700 cm−1).50 The D/G band intensity ratio (ID/IG) is often used for determining the graphene's degree of structural defects.51 Thus, a smaller ID/IG ratio means a lower number of defects and disorders in the graphitic structure, comprising the disorders caused at the edges of the platelets;52 therefore, the D-band intensity is influenced by the flake size.26,53 All prepared graphene films showed ID/IG ratio identical to the one of the pristine graphite ore (ca. 0.15, Table S2†), confirming the high structural integrity of the produced graphene sheets.32
Commercial powdered natural graphite, extracted from the graphite ore by a conventional treatment, was also applied in the developed method and used as a comparison to the direct graphite ore exfoliation. The same pressurized sonication time (30 min) was sufficient to completely delaminate the powdered natural graphite in the biphasic H2O–CH2Cl2 system containing 0.3 wt% of BMImNTf2. The XRD pattern (Fig. S2†), TEM images and SAED (Fig. S5†) gave evidence that few-layers and monolayer graphene sheets were also formed. The similar graphene sheets obtained, for both graphite ore and commercial powdered graphite, evidence the effectiveness of the process and highlight the advantages of a direct processing of the graphite ore.
Most interestingly, when a higher amount of CH2Cl2 (50 v%) was added to the process a different behavior was observed after 30 min of sonication. Macroporous swelled species with micrometric lateral dimension were formed in high yield (Fig. 5a). These seem to be constituted by few layers of crumpled graphene, forming two main types of structure; (i) large macroporous segments (Fig. 5b), and (ii) pine cone-like structures54 formed by few layers of entangled and crumpled graphene (Fig. 5c). These structures were probably created due to the more intensive intercalation of IL–CH2Cl2 in between graphite galleries. As this phenomenon occurred exclusively with the IL–CH2Cl2 system, it seems to be a synergistic solvent-coupling agent effect. In Fig. 5d and e, AFM evidenced the formation of a graphene bilayer (∼1 nm thick) with swollen central regions (2–3 nm thick), indicating that the swelling could be due to cavitation induced bubble formation from the intercalated IL–CH2Cl2. The two different structures formed could be a result of different local sheet size and chemical composition. The phase transition between a flat-membrane phase and the crumpled phase has been reported to have a notable dependence on the size, functionalization and defects of the graphene sheets.55 Thus; the predominant presence of layered crumpled graphene structures was also an indication of the small content of sheet defects.
 | ||
| Fig. 5 Microscopic images of the crumpled graphene–graphite species formed by biphasic pressurized sonication procedure at higher CH2Cl2 content; TEM image the general morphological aspect (a) and higher magnification of the “macroporous” (b) and “pine cone-like” (c) species formed. AFM topological image (d) and height profile (e) of a swelled graphene bilayer. | ||
Conclusions
The pressurized sonication of a biphasic system represents a cheap and effective method for the exfoliation of graphite ore into few-layers graphene. This reaction set-up avoids the need of a centrifugation purification procedure, as the mineral impurities originated from the graphite ore are collected in the water phase or settle down at the bottom, whilst the produced graphene sheets are located at the liquid–liquid interface and in the organic phase. The addition of IL (BMImNTf2) considerably reduced the exfoliation time and produced graphene sheets with much larger lateral dimensions. Although preliminary, also a prospective, simple and straightforward method for the controlled production of crumpled graphene materials has been demonstrated. Since these processes produce graphene sheets with non-covalently bonded BMImNTf2 on the surface, the modified graphene species could be used in a vast range of materials applications without altering the pristine graphene characteristics. The system optimization and the role of different ionic liquids into the final materials' morphology are an active subject of our group and will be explored in details in further publications.Acknowledgements
The authors acknowledge the Grant Agency of the Czech Republic (project 14-05146S) for financial support. R. K. Donato is thankful to FAPERGS-CAPES for the DOCFIX post-doctoral fellowship. H. S. Schrekker is grateful to CNPq for the PQ fellowship. The authors thank Zuzana Bělčická for the Raman measurements.References
- M. Terrones, A. R. Botello-Mendez, J. Campos-Delgado, F. Lopez-Urias, Y. I. Vega-Cantu, F. J. Rodriguez-Macias, A. L. Elias, E. Munoz-Sandoval, A. G. Cano-Marquez, J. C. Charlier and H. Terrones, Nano Today, 2010, 5, 351 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- X. Zhang, A. C. Coleman, N. Katsonis, W. R. Browne, B. J. van Wees and B. L. Feringa, Chem. Commun., 2010, 46, 7539 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- O. C. Compton and S. B. T. Nguyen, Small, 2010, 6, 711 CrossRef CAS PubMed.
- X. Qi, K.-Y. Pu, X. Zhou, H. Li, B. Liu, F. Boey, W. Huang and H. Zhang, Small, 2010, 6, 663 CrossRef CAS PubMed.
- S. Vadahanambi, J.-H. Jung, R. Kumar, H.-J. Kim and I.-K. Oh, Carbon, 2013, 53, 391 CrossRef CAS.
- A. C. Kleinschmidt, R. K. Donato, M. Perchacz, H. Benes, V. Stengl, S. C. Amico and H. S. Schrekker, RSC Adv., 2014, 4, 43436 RSC.
- Q. Liu, T. Fujigaya and N. Nakashima, Carbon, 2012, 50, 5421 CrossRef CAS.
- K. V. Emtsev, A. Bostwick, K. Horn, J. Jobst, G. L. Kellogg, L. Ley, J. L. McChesney, T. Ohta, S. A. Reshanov, J. Rohrl, E. Rotenberg, A. K. Schmid, D. Waldmann, H. B. Weber and T. Seyller, Nat. Mater., 2009, 8, 203 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312 CrossRef CAS PubMed.
- J.-W. Lee, M. Kim, W. Na, S. M. Hong and C. M. Koo, Carbon, 2015, 91, 527 CrossRef CAS.
- A. Dato, V. Radmilovic, Z. Lee, J. Phillips and M. Frenklach, Nano Lett., 2008, 8, 2012 CrossRef CAS PubMed.
- S. Guo and S. Dong, Chem. Soc. Rev., 2011, 40, 2644 RSC.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- V. Stengl, Chem.–Eur. J., 2012, 18, 14047 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- W. F. Zhao, M. Fang, F. R. Wu, H. Wu, L. W. Wang and G. H. Chen, J. Mater. Chem., 2010, 20, 5817 RSC.
- Z. Wei, D. E. Barlow and P. E. Sheehan, Nano Lett., 2008, 8, 3141 CrossRef CAS PubMed.
- Z. Osvath, A. Darabont, P. Nemes-Incze, E. Horvath, Z. E. Horvath and L. P. Biro, Carbon, 2007, 45, 3022 CrossRef CAS.
- E. K. Choi, I. Y. Jeon, S. Y. Bae, H. J. Lee, H. S. Shin, L. M. Dai and J. B. Baek, Chem. Commun., 2010, 46, 6320 RSC.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611 CrossRef CAS PubMed.
- R. Peng, Y. Wang, W. Tang, Y. Yang and X. Xie, Polymers, 2013, 5, 847 CrossRef.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14 CrossRef CAS PubMed.
- S. Wang, Y. Zhang, N. Abidi and L. Cabrales, Langmuir, 2009, 25, 11078 CrossRef CAS PubMed.
- S. Bellayer, J. W. Gilman, N. Eidelman, S. Bourbigot, X. Flambard, D. M. Fox, H. C. de Long and P. C. Trulove, Adv. Funct. Mater., 2005, 15, 910 CrossRef CAS.
- X. Q. Wang, P. F. Fulvio, G. A. Baker, G. M. Veith, R. R. Unocic, S. M. Mahurin, M. F. Chi and S. Dai, Chem. Commun., 2010, 46, 4487 RSC.
- D. Nuvoli, L. Valentini, V. Alzari, S. Scognamillo, S. B. Bon, M. Piccinini, J. Illescas and A. Mariani, J. Mater. Chem., 2011, 21, 3428 RSC.
- N. G. Shang, P. Papakonstantinou, S. Sharma, G. Lubarsky, M. Li, D. W. McNeill, A. J. Quinn, W. Zhou and R. Blackley, Chem. Commun., 2012, 48, 1877 RSC.
- M. Matsumoto, Y. Saito, C. Park, T. Fukushima and T. Aida, Nat. Chem., 2015, 7, 730 CrossRef CAS PubMed.
- M. Peplow, Nature, 2015, 522, 268 CrossRef CAS PubMed.
- B. Sakintuna, Y. Yurum and S. Cetinkaya, Energy Fuels, 2004, 18, 883 CrossRef CAS.
- B. Saner, F. Okyay and Y. Yurum, Fuel, 2010, 89, 1903 CrossRef CAS.
- http://www.asbury.com/technical-presentations-papers/materials-in-depth/natural-flake-graphite/ .
- F. Fenouillot, P. Cassagnau and J. C. Majeste, Polymer, 2009, 50, 1333 CrossRef CAS.
- S. Biswas and L. T. Drzal, Nano Lett., 2008, 9, 167 CrossRef PubMed.
- S. J. Woltornist, A. J. Oyer, J. M. Y. Carrillo, A. V. Dobrynin and D. H. Adamson, ACS Nano, 2003, 7, 7062 CrossRef PubMed.
- T. Fukushima and T. Aida, Chem.–Eur. J., 2007, 13, 5048 CrossRef CAS PubMed.
- X. Zhou, T. Wu, K. Ding, B. Hu, M. Hou and B. Han, Chem. Commun., 2010, 46, 386 RSC.
- B. T. Kelly, Physics of Graphite, Applied Science, London, 1981 Search PubMed.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis and A. K. Stubos, Small, 2009, 5, 1841 CrossRef CAS PubMed.
- P. Nemes-Incze, Z. Osváth, K. Kamaras and L. P. Biro, Carbon, 2008, 46, 1435 CrossRef CAS.
- I. Jung, D. A. Dikin, R. D. Piner and R. Ruoff, Nano Lett., 2008, 8, 4283 CrossRef CAS PubMed.
- M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3, 11700 CAS.
- J. H. Warner, M. H. Rummeli, L. Ge, T. Gemming, B. Montanari, N. M. Harrison, B. Buchner and G. A. D. Briggs, Nat. Nanotechnol., 2009, 4, 500 CrossRef CAS PubMed.
- V. Stengl, J. Henych, J. Bludska, P. Ecorchard and M. Kormunda, Ultrason. Sonochem., 2015, 24, 65 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- R. Satio, M. Hofmann, G. Dresselhaus, A. Jorio and M. S. Dresselhaus, Adv. Phys., 2011, 60, 413 CrossRef.
- M. M. Lucchese, F. Stavale, E. M. Ferreira, C. Vilani, M. V. O. Moutinho, R. B. Capaz, C. A. Achete and A. Jorio, Carbon, 2010, 48, 1592 CrossRef CAS.
- O. Akhavan, ASC Nano, 2010, 4, 4174 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864 CrossRef CAS PubMed.
- Y. Zhao, J. Feng, X. Liu, F. Wang, L. Wang, C. Shi, L. Huang, X. Feng, X. Chen, L. Xu, M. Yan, Q. Zhang, X. Bai, H. Wu and L. Mai, Nat. Comm., 2014, 5, 4565 CAS.
- C. Chang, Z. Song, J. Lin and Z. Xu, RSC Adv., 2013, 3, 2720 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23654a |
| This journal is © The Royal Society of Chemistry 2016 |