DOI:
10.1039/C5RA23568E
(Paper)
RSC Adv., 2016,
6, 5535-5540
Oxygen reduction reaction on cobalt–(n)pyrrole clusters from DFT studies†
Received
9th November 2015
, Accepted 24th December 2015
First published on 6th January 2016
Abstract
The oxygen reduction reaction (ORR) catalyzed by Co–(n)PPy (n = 2–10) clusters is investigated in detail at BLYP/DZP level of theory. The calculation results indicate that different O2 adsorption modes could greatly affect the types of the reduction intermediates. The side-on O2 adsorption is more likely to yield the intermediate HO–OH, while end-on yields H2O–O. However, the side-on O2 adsorption could lead to strong intra-molecular strain and result in instability of the clusters, the cobalt ion therefore is more easily to be dissolved from the active site, leading to poor durability of the Co–(n)PPy clusters. The ORR activity might be enhanced with the cluster size increases, based on HOMO and LUMO analysis. From Co–(8)PPy, the electronic structures are hard to be modified by simply increasing the PPy chains. However, further increasing the cluster size might result in an increase of Co–N2 active site due to that more Co atoms could be captured by the pyrrolic N atoms, the resulting synergistic effect would be more likely to enhance the activity.
1. Introduction
To solve the major obstacle to the commercialization of proton exchange membrane fuel cells (PEMFCs), that is, the high cost and the low stability of Pt-based materials, many efforts have been made to develop cheap, durable and more active non-Pt electrocatalysts. To date, the most promising non-precious metal electrocatalysts for PEMFCs are carbon-supported transition metal/nitrogen (M–Nx/C) materials (M = Co, Fe), which have gained increasing attention due to their promising catalytic performance towards the oxygen reduction reaction (ORR). This class of electrocatalysts are usually synthesized by the simple pyrolysis of transition metal, carbon and some nitrogen containing materials, such as macrocycle compounds, organic complexes, nitrogen containing salts and even gaseous nitrogen precursors.1
Due to the chemical structure of the precursors during high temperature decomposition, the active site structure is not yet clearly understood, making the actual nature of the active sites being a subject of debate.2 For example, some species, such as M–N4/C,3,4 M–N2/C5 and N–C6,7 have been proposed as the catalytic active sites. The different reports above could be mostly attributed to differences in the synthesis procedures and the nature of the characterization techniques utilized.1 Obviously, these conflicting results hinder the understanding of the ORR mechanism and block the designing of more efficient M–Nx/C electrocatalysts. Non-pyrolyzed M–Nx/C catalysts on the other hand have attracted interest because their well-defined structures are preserved during simple synthesis process, providing the ability to detect their active sites and catalytic mechanisms.
Transition metal doped conductive polymer materials, for instance, the cobalt–polypyrrole (Co–PPy) complexes,8–12 have shown their great potential in the electrocatalytic ORR processes. The non-pyrolyzed Co–PPy electrocatalyst reported by Bashyam and Zelenay exhibited a high ORR activity,8 without showing any noticeable loss of performance during operation of the PEMFC. The observed ORR activity was attributed to the strong Co–PPy interactions, and assumed to form the Co–N catalytic active sites. Furthermore, the chemical valence of Co is considered to be +2 according to the 2p1/2 and 2p2/3 electron binding energies of cobalt.13 Based on these experimental results, some theoretical works focus on the structures of the active site, devoting to clarify the mechanism of ORR and shed light on the origin of the electrocatalytic activity.14–18 These studies gave us a better understanding of the catalytic details, which are not able to detect from the experiments. For example, the Co–N2 active site structures and the platinum-like catalytic behavior could be found according to the theoretical works above. However, some questions remain unclear, especially regarding to the reduction mechanism: why do experimental studies on the Co–PPy systems have shown that both two-electron and four-electron ORR mechanism are observed? Whether the cluster size influences the adsorption of ORR intermediates, thereby affecting the mechanism and activity? All these questions need further investigation to clarify the observed catalytic behavior.
In this paper, the ORR mechanisms catalyzed by Co–(n)PPy (n = 2–10) were investigated in detail by means of DFT method. The interactions between O2 and Co–(n)PPy, which are identified as end-on and side-on adsorption modes, are both simulated. The calculation results indicate that different adsorption modes could greatly affect the types of the reduction intermediates. Furthermore, with the cluster size increases, the activity as well as the durability are both improved for the studied Co–(n)PPy clusters.
2. Computational methods
DFT calculations were performed by means of the Amsterdam Density Functional program package (ADF).19 The BLYP functional was used to describe the electronic interaction,20 which is commonly used for electrochemical systems.21 The cobalt atom was calculated with a triple-ζ polarized (TZP) slater-type basis set while other atoms were based on double-ζ polarized (DZP) set. Mixing basis set of different sizes was successfully applied on such systems,22 and the basis set effect was demonstrated to be very small.23 The inner core orbitals, 1s for C, N, and O and 1s–3p for Co, were kept frozen. All atoms were allowed to relax without constraint during the geometry optimizations.
The adsorption energy of the ORR intermediates is an important reference point for determining the activity and durability of an electrocatalyst. In this paper, the adsorption energy of the ORR species is defined as the energy difference between the adsorption and the isolated systems. For example, the adsorption energy of molecular O2 is defined as ΔE(O2) = E(Co–PPy–O2) − E(Co–PPy) − E(O2). Therefore, the negative adsorption energy suggests that the O2 molecule would be energetically favorable to be adducted to the Co–(n)PPy clusters.
3. Results and discussion
3.1. The structural properties of Co–(n)PPy clusters
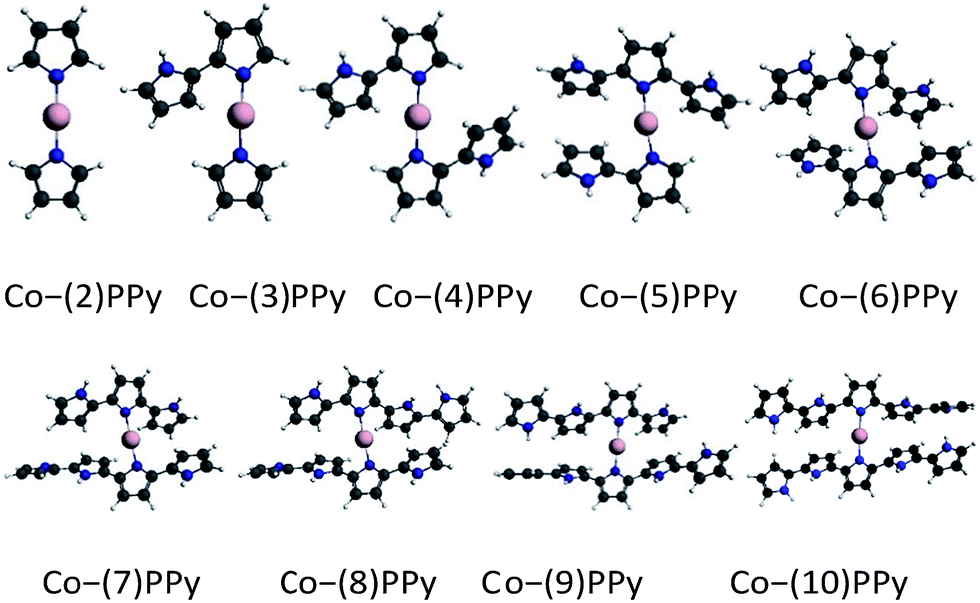
The optimized Co–(n)PPy structures in the studied range are all shown in Fig. 1, where the Co atom is bonded to two N atoms to form the Co–N2 active sites. Furthermore, the calculated structural properties for the above clusters are given in Table 1. From the table, one can clearly find that the bond length of Co–N for the smaller Co–(n)PPy (n < 5) clusters is slightly shorter than that in the larger clusters (n > 5). Meanwhile, the bond angle of N(2)Co(1)N(3) (atomic numbers can be seen in Fig. 2) has an obvious change when n > 5. The Co–N bond length in Co–(n)PPy (n > 5) is around 1.95 Å, which is more consistent with the experimentally determined value of 1.94 (±0.02) Å.11 Because of the high rotational degree of freedom along the Co–N bond, all the configurations of optimized Co–(n)PPy are non-planar, even for the smallest cluster. With the cluster size increases, the steric hindrance of the pyrrole rings becomes more obvious, which would interfere the adsorption mode of the O2 molecule (we will discuss it below).
 |
| | Fig. 1 Optimized structures of Co–(n)PPy clusters. The configuration of Co–(10)PPy can also be found in ref. 14. | |
Table 1 Calculated key bond lengths, R (Å), and bond angles, θ (deg), for the studied Co–(n)PPy clusters (the atomic numbers can be found in Fig. 2)
| Co–(n)PPy |
RCo(1)–N(2) |
RCo(1)–N(3) |
θN(2)Co(1)N(3) |
| Co–(2)PPy |
1.892 |
1.892 |
178.9 |
| Co–(3)PPy |
1.928 |
1.893 |
177.4 |
| Co–(4)PPy |
1.921 |
1.92 |
179.3 |
| Co–(5)PPy |
1.934 |
1.909 |
172.9 |
| Co–(6)PPy |
1.952 |
1.953 |
161.4 |
| Co–(7)PPy |
1.945 |
1.952 |
162.1 |
| Co–(8)PPy |
1.948 |
1.944 |
165 |
| Co–(9)PPy |
1.946 |
1.95 |
174 |
| Co–(10)PPy |
1.95 |
1.955 |
176.1 |
 |
| | Fig. 2 Optimized end-on and side-on O2 adsorption modes on the studied Co–(n)PPy clusters. | |
3.2. Adsorption of oxygen molecule
As is well known, the adsorption of O2 is the first step for the ORR, and the adsorption energy is important to determine the catalytic activity. In current work, two O2 adsorption modes, end-on and side-on are both calculated and their structures are fully optimized, as shown in Fig. 2. Furthermore, the calculated adsorption properties, such as the adsorption energies, key bond lengths and bond angles for Co–(n)PPy–O2 complexes are listed in Table 2. The O2 adsorption energy of the side-on mode for all the studied Co–(n)PPy clusters is in the range of −1.11 to −1.33 eV, which is about 0.45–0.65 eV stronger than their corresponding end-on mode. However, the experiment determined the low-coverage adsorption energy of O2 on Pt(111) to be −0.3 to −0.5 eV,24,25 and theoretically determined to be −0.41 to −1.04 eV, based on different methods and models.26–29 In general the adsorption energy of O2 on an effective electrocatalyst must be within a certain range, too strong or too weak are all unfavorable for the ORR. In other words, we might think that the O2 adsorption energy on an ideal catalytic material should be as small as possible, but large enough to prevent O2 from drifting away or desorbing from the catalytic center.25 Obviously, the side-on O2 adsorption on the Co–(n)PPy clusters is too strong, which would block the subsequent oxygen reduction steps. Furthermore, for a larger Co–(n)PPy (n > 5), because of the side-on adsorption needs more space than that of end-on mode, the PPy chains are significantly rotated and become perpendicular to each other, as shown in Fig. 2. These cross-linked PPy rings significantly changes the initial structures of the Co–(n)PPy, which is also demonstrated from Table 2.
Table 2 Calculated key bond lengths, R (Å), bond angles, θ (deg), and end-on O2 adsorption energies, ΔE (eV), for the Co–(n)PPy–O2 complexes (the data corresponding to the side-on adsorption mode are given in the parentheses)
| Co–(n)PPy |
RCo(1)–N(2) |
RCo(1)–N(3) |
RCo(1)–O(4) |
RCo(1)–O(5) |
RO(4)–O(5) |
θN(1)Co(2)N(3) |
θCo(1)O(4)O(5) |
ΔE |
| Co–(2)PPy |
1.877 |
1.876 |
1.795 |
2.57 |
1.311 |
132.8 |
110.7 |
−0.71 |
| (1.962) |
(1.965) |
(1.831) |
(1.832) |
(1.425) |
(91.7) |
(67.1) |
(−1.26) |
| Co–(3)PPy |
1.907 |
1.882 |
1.785 |
2.644 |
1.308 |
130.6 |
116.7 |
−0.68 |
| (2.005) |
(1.927) |
(1.862) |
(1.808) |
(1.423) |
(94.1) |
(65.2) |
(−1.31) |
| Co–(4)PPy |
1.908 |
1.904 |
1.777 |
2.739 |
1.309 |
128.4 |
122.9 |
−0.65 |
| (1.957) |
(1.999) |
(1.819) |
(1.857) |
(1.419) |
(101.6) |
(68.7) |
(−1.14) |
| Co–(5)PPy |
1.973 |
1.976 |
1.786 |
2.62 |
1.326 |
146.2 |
113.9 |
−0.67 |
| (2.011) |
(1.953) |
(1.849) |
(1.85) |
(1.392) |
(96.7) |
(67.9) |
(−1.11) |
| Co–(6)PPy |
1.972 |
1.993 |
1.79 |
2.612 |
1.321 |
155.7 |
113.3 |
−0.6 |
| (2.01) |
(2.011) |
(1.836) |
(1.84) |
(1.414) |
(106.4) |
(67.6) |
(−1.11) |
| Co–(7)PPy |
1.98 |
1.982 |
1.796 |
2.648 |
1.319 |
152.9 |
115.6 |
−0.58 |
| (2.025) |
(2.025) |
(1.865) |
(1.862) |
(1.391) |
(105.2) |
(68) |
(−1.13) |
| Co–(8)PPy |
1.997 |
1.991 |
1.795 |
2.644 |
1.34 |
152 |
114.2 |
−0.84 |
| (1.986) |
(2.01) |
(1.847) |
(1.843) |
(1.423) |
(99) |
(67.1) |
(−1.33) |
| Co–(9)PPy |
2.008 |
1.993 |
1.795 |
2.629 |
1.325 |
164.4 |
114 |
−0.64 |
| (2.016) |
(2.025) |
(1.858) |
(1.859) |
(1.396) |
(106.4) |
(68) |
(−1.23) |
| Co–(10)PPy |
2.005 |
1.996 |
1.79 |
2.603 |
1.329 |
163.8 |
112.3 |
−0.71 |
| (2.004) |
(2.022) |
(1.839) |
(1.831) |
(1.417) |
(106.8) |
(67) |
(−1.22) |
Also from Table 2, one can easily find that for each Co–(n)PPy cluster, almost all the Co–N bond length have been elongated after O2 adsorption, accompanied by a decrease of N(2)Co(1)N(3) bond angle. Note that for side-on adsorption configuration, the variation of the above parameters are more significant than the corresponding end-on mode. All the data above indicate that the side-on O2 adsorption could lead to a strong intra-molecular strain, which suggests an instability of the clusters. Therefore, for the side-on configuration, the cobalt atom is more easily dissolved from the active site, leading to a poor durability of the Co–(n)PPy clusters. On the contrary, the end-on mode has relative weaker O2 adsorption energies, only resulting in a smaller structural distortion from the initial catalytic configuration, which makes it favorable for the subsequent ORR.
3.3. Effect of O2 adsorption mode on the ORR mechanism
For each Co–(n)PPy cluster, the O–O bond length of side-on O2 adsorption is about 0.1 Å longer than that of end-on mode, as shown in Table 2. This is attributed to the greater d–π* orbital overlapping between cobalt and O2 for side-on adsorption.17 Yeager has proposed that the type of interaction between catalyst and O2 molecule dictates the oxygen reduction pathway.30 Normally, the side-on adsorption is more likely to lead to a 4e− ORR due to more electrons could be transferred to O2, while the end-on adsorption is easier to result in a 2e− ORR to H2O2. In this paper, the ORR on Co–(n)PPy clusters is investigated in detail, and each step can be summarized as follows:| | |
*OOH + H+ + e− → *H2O–O (or *HO–OH)
| (3-1) |
| | |
*OOH + H+ + e− → *H2O2
| (3-2) |
| | |
*H2O–O + H+ + e− → *OH + H2O
| (4-1) |
| | |
overall: O2 + 4H+ + 4e− → 2H2O
| (7) |
where the asterisk represents a chemisorption site on Co–(n)PPy clusters. The reaction eqn (3), that is, the second electron transfer is the key step to determine the ORR mechanism. The optimized configurations for the reaction products of the second electron transfer are shown in Fig. S1,† and the calculated data are listed in Table S1.† We find that for both end-on and side-on O2 adsorption modes, the O–O bond is completely broken after the second electron transfer step, indicating the 4e− ORR pathway. However, the side-on O2 adsorption is more likely to yield the intermediate HO–OH (two adsorbed hydroxyl groups), while end-on yields H2O–O (one adsorbed atomic oxygen and one water molecule). From Table S1,† one can find that yielding HO–OH intermediate could result in a large structural distortion relative to the initial Co–(n)PPy structures, which is similar to the situation that observed in Co–(n)PPy–O2 complexes (side-on adsorption configurations).
The obtained 4e− ORR mechanism is consistent with some previous experimental and theoretical works,8,14,15,18 where they demonstrated that the O2 could be directly reduced to H2O on Co–N2 active site, with the generation of H2O2 is negligible. While some studies also show that the Co–PPy could only catalyze 2e− or incomplete 4e− ORR reaction pathways, especially for pyrolyzed samples.10,11 Also note that a different result reported by Zhang et al. have shown that pyrolysis could result in activity enhancement as well as mechanism change from a 2e− dominant to a 4e− dominant reduction process,31 which makes more difficult to understand the nature of observed catalytic behavior. As is well known, the electrochemical ORR on catalysts is very complex, it is influenced by many factors such as the catalyst synthesis condition, surface structure, structure size, potential, and liquid environment, etc. For instance, the catalytic mechanism is sensitive to the operation potentials. The Co–N2 active site, which is investigated in the current paper, could catalyze 4e− ORR, whether at high or low potentials, while others not.15 The observed 2e− or incomplete 4e− ORR pathways on pyrolyzed Co–PPy may attributed to other active sites (or mixed sites), such as Co–N3.18 Furthermore, due to the fact that the calculated one-unit Co–(2)PPy is the least networked structure, it is unlikely to exist after the high-temperature treatment, which will lead to a reduction in the H2O2 yield.15 Such case was indeed observed in experimental work.31
Fig. 3 shows the respective changing ratios of bond parameters from Co–(n)PPy–O2 (end-on adsorption) to Co–(n)PPy–H2O–O, and Co–(n)PPy–O2 (side-on adsorption) to Co–(n)PPy–HO–OH configurations. We find that all the changing ratios for bond lengths are in the range of less than 8%, and for bond angles less than 25%, indicating there are no obvious structural changes during these steps. Thus, the types of intermediates are strongly affected by the O2 adsorption mode, that is, the end-on adsorption corresponds to the H2O–O while the side-on corresponds to the HO–OH. However, yielding HO–OH would also make Co–(n)PPy a poor durability due to the large structural distortion relative to the initial structures, as mentioned above.
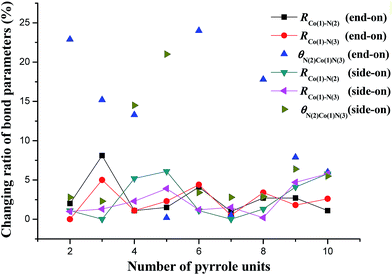 |
| | Fig. 3 The changing ratios of bond parameters from Co–(n)PPy–O2 (end-on adsorption) to Co–(n)PPy–H2O–O, and Co–(n)PPy–O2 (side-on adsorption) to Co–(n)PPy–HO–OH configurations. | |
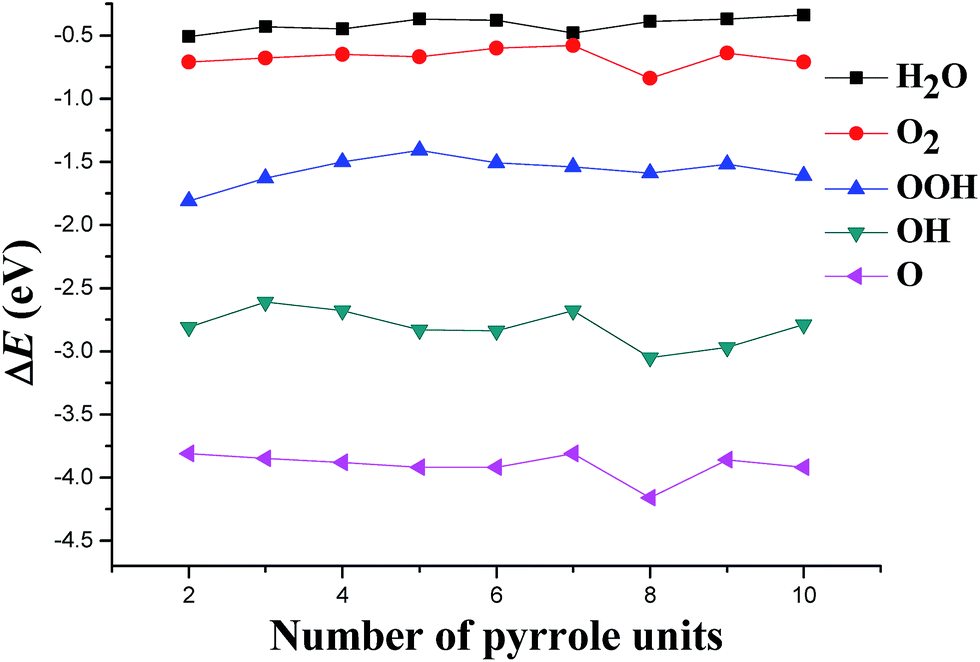
3.4. Adsorption of ORR species
Fig. 4 gives the adsorption energies of all species involved in the ORR. Generally, a complete catalytic cycle of ORR starts from the O2 adsorption, and ends to the H2O desorption. Therefore, for an effective ORR catalyst, the H2O molecule would coordinate more weakly to it than to the others, such as the O2, so that the catalytic cycle could repeat most easily for it.32 From Fig. 4, it can be find that the adsorption energy differences between H2O and O2 have smaller changes before Co–(7)PPy, but from Co–(8)PPy, the changes become bigger. Therefore, a larger Co–(n)PPy cluster looks like more favorable for the ORR investigated.
 |
| | Fig. 4 Adsorption energies of the oxygen reduction species on the Co–(n)PPy clusters (the adsorption energy of O2 is related to the end-on mode). | |
With the cluster size increases, the adsorption energies of OOH have been decreased, but from Co–(6)PPy, the values gradually stabilized to about −1.5 eV. The adsorption energies of atomic O on all Co–(n)PPy clusters have no bigger changes, but for OH, the situation is quite different. For example, for the larger clusters, the adsorption energies decrease in the sequence Co–(8)PPy > Co–(9)PPy > Co–(10)PPy. The decreased OH adsorption energies would result in a decrease in the overpotential required for OH reduction, which is also demonstrated experimentally on the Pt-based alloy materials.33
3.5. Cluster size effect
Previous studies suggested that the size effect is an important factor to affect the catalytic activity,34,35 since the electronic structure could be fluctuated with the cluster size variation. The energy levels of the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO) and the highest occupied 3d orbital of Co (named as Co-3d) are listed in Table 3. We find that with the cluster size increases, the energy levels of HOMO (as well as Co-3d) and LUMO are both enhanced, and the values of HLG (energy gap between HOMO and LUMO) almost decreases simultaneously. This indicates that the ORR activity would be enhanced with the cluster size increases. However, this tendency becomes slow after Co–(7)PPy, which suggests that the electronic structures are no more modified by increasing the PPy chains. Nevertheless, in a larger cluster with longer PPy chains, more Co atoms could be captured by the pyrrolic N atoms to form Co–N2 active site, just like the situation in our previous work.14 In that case the synergistic effect between Co atoms may result in higher catalytic activity.
Table 3 HOMO, Co-3d, LUMO, and HLG for all the studied clusters (unit: eV)a
| Co–(n)PPy |
HOMO |
LUMO |
Co-3d |
HLG |
| HLG is the energy gap between HOMO and LUMO energy levels. |
| Co–(2)PPy |
−5.372 |
−5.094 |
−6.059 |
0.278 |
| Co–(3)PPy |
−5.048 |
−4.501 |
−5.527 |
0.547 |
| Co–(4)PPy |
−4.74 |
−4.353 |
−5.386 |
0.387 |
| Co–(5)PPy |
−4.518 |
−4.232 |
−5.156 |
0.286 |
| Co–(6)PPy |
−4.31 |
−4.066 |
−4.407 |
0.244 |
| Co–(7)PPy |
−4.241 |
−3.985 |
−4.332 |
0.255 |
| Co–(8)PPy |
−4.206 |
−3.977 |
−4.291 |
0.229 |
| Co–(9)PPy |
−4.219 |
−4.082 |
−4.219 |
0.137 |
| Co–(10)PPy |
−4.191 |
−4.054 |
−4.223 |
0.137 |
4. Conclusions
In summary, the ORR on Co–(n)PPy (n = 2–10) clusters is investigated in detail at BLYP/DZP level of theory. The following conclusions can be drawn from this work:
(1) The side-on O2 adsorption configuration is more stable than the corresponding end-on mode for all the investigated Co–(n)PPy clusters, but the adsorption for the former is stronger when compared with that on Pt surface, which would block the subsequent oxygen reduction steps. The end-on configuration has the appropriate adsorption energy and is favorable for ORR.
(2) The side-on O2 adsorption could lead to a strong intra-molecular strain and result in an instability of the clusters. In that case the cobalt atom is more easily dissolved from the active site, leading to a poor durability of the Co–(n)PPy clusters.
(3) The adsorption mode of O2 determines the reduction intermediates. The end-on mode is more likely to yield the intermediate of H2O–O, while side-on yields HO–OH. But once again, yielding HO–OH could lead to large structural distortion relative to the initial Co–(n)PPy structures and thus result in poor durability.
(4) For the larger clusters, the OH adsorption energies decrease in the sequence Co–(8)PPy > Co–(9)PPy > Co–(10)PPy. The decreased OH adsorption would result in a decrease in the overpotential required for the OH reduction, which is also demonstrated experimentally on the Pt-based alloy materials.
(5) The ORR activity might be enhanced with the cluster size increases, based on analysis of the energy levels of HOMO and LUMO. However, from Co–(8)PPy, the electronic structures are hard to be modified by simply increasing the PPy chains.
Acknowledgements
The author thank the Supercomputing Center of the Chinese Academy of Sciences and Beijing University of Technology for providing the computational resources and software.
Notes and references
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 Search PubMed.
- W. Li, A. Yu, D. C. Higgins, B. G. Llanos and Z. Chen, J. Am. Chem. Soc., 2010, 132, 17056–17058 CrossRef CAS PubMed.
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CAS.
- U. Kramm, I. Abs-Wurmbach, I. Herrmann-Geppert, J. Radnik, S. Fiechter and P. Bogdanoff, J. Electrochem. Soc., 2011, 158, B69–B78 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef.
- G. Liu, X. G. Li, P. Ganesan and B. N. Popov, Electrochim. Acta, 2010, 55, 2853–2858 CrossRef CAS.
- Y. Nabae, S. Moriya, K. Matsubayashi, S. M. Lyth, M. Malon, L. B. Wu, N. M. Islam, Y. Koshigoe, S. Kuroki, M. A. Kakimoto, S. Miyata and J. Ozaki, Carbon, 2010, 48, 2613–2624 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- T. S. Olson, S. Pylypenko and P. Atanassov, J. Phys. Chem. C, 2010, 114, 5049–5059 CAS.
- W. Feng, H. Li, X. Cheng, T. C. Jao, F. B. Weng, A. Su and Y. C. Chiang, Appl. Surf. Sci., 2012, 258, 4048–4053 CrossRef CAS.
- D. Nguyen-Thanh, A. I. Frenkel, J. Wang, S. O, Brien and D. L. Akins, Appl. Catal., B, 2011, 105, 50–60 CrossRef CAS.
- C. Walter, K. Kummer, D. Vyalikh, V. Brüser, A. Quade and K. D. Weltmann, J. Electrochem. Soc., 2012, 159, F494–F500 CrossRef CAS.
- X. Wang, J. Xu, B. Zhang, H. Yu, J. Wang, X. Zhang, J. Yu and Q. Li, Adv. Mater., 2006, 18, 2476–2480 CrossRef CAS.
- X. Chen, F. Li, X. Wang, S. Sun and D. Xia, J. Phys. Chem. C, 2012, 116, 12553–12558 CAS.
- Z. Shi, H. Liu, K. Lee, E. Dy, J. Chlistunoff, M. Blair, P. Zelenay, J. Zhang and Z. S. Liu, J. Phys. Chem. C, 2011, 115, 16672–16680 CAS.
- H. K. Dipojono, A. G. Saputro, S. M. Aspera and H. Kasai, Jpn. J. Appl. Phys., 2011, 50, 055702 CrossRef.
- H. K. Dipojono, A. G. Saputro, R. Belkada, H. Nakanishi, H. Kasai, M. David and E. S. Dy, J. Phys. Soc. Jpn., 2009, 78, 094710 CrossRef.
- A. G. Saputro and H. Kasai, J. Phys. Soc. Jpn., 2014, 83, 024707 CrossRef.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. J. Ziegler, Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- X. Chen, Q. Qiao, L. An and D. Xia, J. Phys. Chem. C, 2015, 119, 11493–11498 CAS.
- C. Dragonetti, S. Righetto, D. Roberto, R. Ugo, A. Valore, S. Fantacci, A. Sgamellotti and F. D. Angelis, Chem. Commun., 2007, 40, 4116–4118 RSC.
- A. Michalak, R. L. DeKock and T. Ziegler, J. Phys. Chem. A, 2008, 112, 7256–7263 CrossRef CAS PubMed.
- J. A. Keith and T. Jacob, Angew. Chem., Int. Ed., 2010, 49, 9521–9525 CrossRef CAS PubMed and references therein.
- A. Lyalin, A. Nakayama, K. Uosaki and T. Taketsugu, Phys. Chem. Chem. Phys., 2013, 15, 2809–2820 RSC.
- Y. Sha, T. H. Yu, Y. Liu, B. V. Merinov and W. A. Goddard III, J. Phys. Chem. Lett., 2010, 1, 856–861 CrossRef CAS.
- L. Qi, X. Qian and J. Li, Phys. Rev. Lett., 2008, 101, 146101 CrossRef PubMed.
- M. P. Hyman and J. W. Medlin, J. Phys. Chem. C, 2007, 111, 17052–17060 CAS.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X. F. Zhou, H. T. Wang, J. J. Xu and E. G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155454 CrossRef.
- E. Yeager, J. Mol. Catal., 1986, 38, 5–25 CrossRef CAS.
- K. Lee, L. Zhang, H. Liu, R. Hui, Z. Shi and J. Zhang, Electrochim. Acta, 2009, 54, 4704–4711 CrossRef CAS.
- E. Vayner and A. B. Anderson, J. Phys. Chem. C, 2007, 111, 9330–9336 CAS.
- I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen, J. Rossmeisl and I. Chorkendorff, J. Am. Chem. Soc., 2011, 133, 5485–5491 CrossRef CAS PubMed.
- B. C. Han, C. R. Miranda and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075410 CrossRef.
- D. Deng, L. Yu, X. Pan, S. Wang, X. Chen, P. Hu, L. Sun and X. Bao, Chem. Commun., 2011, 47, 10016–10018 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra23568e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.