
Photoluminescence tuning via energy transfer in Eu-doped Ba2(Gd,Tb)2Si4O13 solid-solution phosphors
Abstract
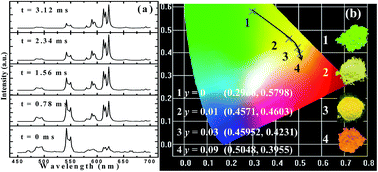
We report on the phase formation of a Ba2(Gd,Tb)2Si4O13 solid-solution, and the coexistence of Eu2+/Eu3+ was identified after Eu ion doping although the samples were prepared in a reducing atmosphere. Under 377 nm near-ultraviolet (UV) light excitation, Ba2Tb2Si4O13 exhibits the characteristic emission originating from Tb3+ corresponding to 5D4–7F6,5,4,3 transitions; whereas Ba2Tb2Si4O13:Eu emits bright red emission from Eu3+ with peaks around 594, 613 and 623 nm. Accordingly, photoluminescence tuning of Eu-doped Ba2(Gd,Tb)2Si4O13 phosphors has been realized from green, yellow, orange, to red emission light. Decay time and time-resolved luminescence results revealed that the tunable luminescence behavior should be ascribed to the existence of energy migration in the terbium subset, and successive energy transfer processes Eu2+–Eu3+(Tb3+) and Tb3+–Eu3+ appear to occur in the Ba2Tb2−ySi4O13:yEu (y = 0–0.12) solid-solution phosphors under investigation.
 Please wait while we load your content...
Please wait while we load your content...

