
Chemical fragment-based CDK4/6 inhibitors prediction and web server†
Abstract
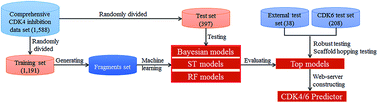
Cyclin-dependent kinases (CDKs), a family of mammalian heterodimeric kinases, play central roles in the regulation of cell cycle progression, transcription, neuronal differentiation, and metabolism. Among these CDKs, CDK4 and CDK6 are considered to be highly validated anticancer drug targets due to their essential role in regulating cell cycle progression at the G1 restriction point. In this study, in silico models derived from 1588 diverse CDK4 enzyme assay data were developed using three machine learning methods, including naïve Bayesian (NB), single tree (ST), and random forest (RF). A total of 1006 models were constructed based on chemical fragments, including fingerprints and atom center fragments (ACFs). The overall predictive accuracies of the best models exceeded 90% for both the training and test sets. The scaffold-hopping abilities of the best models were successfully evaluated by the external test set. Moreover, in silico model-derived CDK4 assay data can predict CDK6 inhibitors (overall predictive accuracies of ∼70–86%). Among all the 1006 models, ACF-based models showed the best performance. A web server was developed based on ACFs and the Bayesian method to predict or design new CDK4/6 inhibitors (CDK4/6 predictor, http://rcidm.org/cdk/).
 Please wait while we load your content...
Please wait while we load your content...

