
Controlling the corrosion rate and behavior of biodegradable magnesium by a surface-immobilized ultrathin 1-hydroxyethylidene-1,1-diphosphonic acid (HEDP) film
Abstract
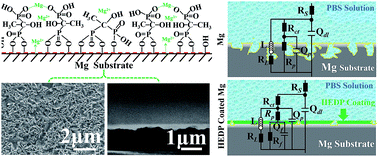
An ultrathin bisphosphonate film, 1-hydroxyethylidene-1,1-diphosphonic acid (HEDP), was deposited on magnesium for biodegradable implant applications. The small, bioactive HEDP molecule is supposed to be not only bio-safe, but also favorable for creating a highly protective layer for the control of the corrosion/degradation of Mg. In an in situ chemical sequence, the HEDP molecules were covalently surface-immobilized on the alkaline pretreated Mg and then spontaneously deposited by participation in a chelating reaction with Mg ions. An organometallic-like compound layer was thus formed, which was ascertained to be within the nanoscale and complied well with the substrate. The tape test showed that the HEDP film provides excellent adhesion strength. Electrochemical corrosion and in vitro immersion degradation investigations demonstrated that the HEDP coated Mg exhibited significantly slower corrosion rate than untreated Mg in phosphate buffered saline (PBS) solution. Of particular significance is the observation that HEDP coated Mg presented a remarkably suppressed localized corrosion mode. The meliorated corrosion/degradation behavior is credited to both the nature of the organometallic-like HEDP derivative layer, as well as the high quality of the film, with respect to compactness and homogeneity. Our HEDP modified Mg may bode well for application in biodegradable implants.
 Please wait while we load your content...
Please wait while we load your content...

