
Activated dopamine derivatives as primers for adhesive-patch fixation of bone fractures†
Abstract
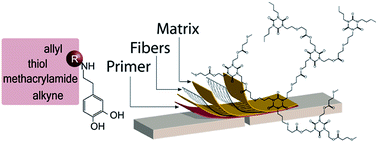
For the stabilization of complex bone fractures, tissue adhesives are an attractive alternative to conventional implants, often consisting of metal plates and screws whose fixation may impose additional trauma on the already fractured bone. This study reports on the synthesis and evaluation of activated dopamine derivatives as primers for fiber-reinforced-adhesive patches in bone-fracture stabilization strategies. The performance of synthesized dopamine derivatives are evaluated with regard to the adhesive shear strength of formed bone patches, as well as cell viability and surface properties. Dopamine-derived primers with methacrylamide, allyl, and thiol functional groups were found to significantly increase the adhesive shear strength of adhesive patches. Furthermore, deprotonation of the primer solution was determined to be essential in order to achieve good adhesion. In conclusion, the primer solutions that were found to give the best adhesion were the once where dopa-thiol was used in combination with either dopa-methacrylamide or dopa-allyl, resulting in shear bond strengths of 0.29 MPa.
 Please wait while we load your content...
Please wait while we load your content...

