
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical mechanistic investigation of zinc(II) catalyzed oxidation of alcohols to aldehydes and esters†
Riffat Un
Nisa
a,
Tariq
Mahmood
a,
Ralf
Ludwig
bc and
Khurshid
Ayub
*a
aDepartment of Chemistry, COMSATS Institute for Information Technology, Campus Abbottabad, Abbottabad, Pakistan. E-mail: khurshid@ciit.net.pk; Fax: +92-992-383441
bLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
cDepartment of Physical Chemistry, University of Rostock, Dr-Lorenz-Weg 1, 18059 Rostock, Germany
First published on 29th March 2016
Abstract
The mechanism of the Zn(II) catalyzed oxidation of benzylic alcohol to benzaldehyde and ester by H2O2 oxidant was investigated through density functional theory methods and compared with the similar oxidation mechanisms of other late transition metals. Both inner sphere and intermediate sphere mechanisms have been analyzed in the presence and absence of pyridine-2-carboxylic acid (ligand). An intermediate sphere mechanism involving the transfer of hydrogen from alcohol to H2O2 was found to be preferred over the competitive inner sphere mechanism involving β-hydride elimination. Kinetic barriers associated with the intermediate sphere mechanism are consistent with the experimental observations, suggesting that the intermediate sphere mechanism is a plausible mechanism under these reaction conditions. The oxidation of alcohols to aldehydes (first step) is kinetically more demanding than the oxidation of hemiacetals to esters (second step). Changing the oxidant to tert-butyl hydrogen peroxide (TBHP) increases the activation barrier for the oxidation of alcohol to aldehyde by 0.4 kcal mol−1, but decreases the activation barrier by 3.24 kcal mol−1 for oxidation of hemiacetal to ester. Replacement of zinc bromide with zinc iodide causes the second step to be more demanding than the first step. Pyridine-2-carboxylic acid ligand remarkably decreases the activation barriers for the intermediate sphere pathway, whereas a less pronounced inverse effect is estimated for the inner sphere mechanism.
Introduction
Esters represent an important class of organic compounds and find applications in fine and bulk chemical industries, fragrances, essential oils, pheromones, pharmaceuticals, agrochemicals,1 plastics and textile industries.2 Conventional methods3–5 of preparing esters generally involve tedious procedures and produce toxic wastes.6 Over the time, several sophisticated strategies have appeared in the literature to build the ester bond, mainly through transition metal catalysis.7–9Recently, palladium catalyzed direct oxidative cross esterification of benzylic and aliphatic alcohols to esters was reported independently by Beller10 and Lei.11 In the direct oxidative cross reaction, oxygen was used as an oxidant and esters were prepared in good yields. The reaction involves the oxidation of alcohol to aldehyde and the resulting aldehyde then reacts with another molecule of alcohol to form the hemiacetal, which on further oxidation delivers the ester product. The reaction is environmentally benign with high atom economy without the need for any activating agent. The concept of oxidation (dehydrogenation) of alcohols is more than 100 years old (Guerbet Chemistry)12 but its elegant use in ester13 and amide14 formation has not been realized until recently. Several other late transition metals have been used for similar oxidation reactions.10,11,13,15–24
Because the catalysts used in the oxidative transformation (mostly Pd, Ir, Ru, and Au) are expensive, the reactions are not economically viable, particularly on an industrial scale. An important factor in the large scale utilization of these reactions is the cost of the catalyst; therefore, recent study is more focused towards the exploration of cheap, non-toxic and environmentally benign catalysts, particularly those based on bio-relevant metals such as iron, zinc, and copper. In this regard, Yefeng Zhu et al. have reported a copper metal based catalyst for the selective oxidation of alcohols to esters, and the selectivity was further extended to benzylic alcohols in DMF.25 More recently, alcohol oxidation to aldehyde and ester through zinc(II) catalyst has also been reported (Scheme 1), where H2O2 is used as the terminal oxidant.26
 | ||
| Scheme 1 Schematic presentation of Zinc catalyzed oxidation of alcohols to esters (ligand (L) = 4–8) | ||
Zinc catalysis, in general, is not well explored, compared to other transition metal catalysis;27 however, a shift in the trend has been observed recently. The importance of zinc catalysis has been demonstrated in several recent reports, including oxidative transformations. The oxidation of alcohols to aldehydes and esters is achieved with zinc bromide and H2O2 as catalyst and oxidant, respectively.26 In a similar approach, aromatic aldehydes have been converted to esters through zinc catalysis.28 Zinc mediated oxidation of ether to aldehydes is also an interesting transformation.29 Although experimental reports have started to emerge, the literature reveals only a handful of theoretical mechanistic studies on zinc catalyzed reactions. In particular, there have been no theoretical reports on the mechanism of any Zn(II) mediated oxidation reactions. A theoretical mechanistic study of the zinc(II) catalyzed oxidation of alcohols to aldehydes and esters under H2O2 and pyridine-2-carboxylic acid ligand is presented here. The main interest behind this study is to compare the mechanism of the zinc catalyzed reaction with other metals (ruthenium and other related metals).
Computational methods
All calculations were performed with Gaussian 09 software.30 Geometries of the structures were optimized without any symmetry constraints at hybrid B3PW91, using the 6-311G(d,p)31 basis set for C, H, N and oxygen, and SDDALL pseudopotential32 for Zn, Br and I, unless otherwise noted. The B3PW91 method, which consists of a three parameters hybrid functional developed by Becke,33 in conjunction with the gradient corrected correlation functional of Perdew and Wang,34 was chosen because it has been shown to reliably model a similar oxidative reaction using Ru metal.35 Each optimized structure was confirmed by frequency analysis at the same level to confirm the stationary point as a true minimum (no imaginary frequency) or a transition state (with one imaginary frequency). Intrinsic reaction coordinate (IRC) calculations were performed to confirm that the transition states connect to the right starting materials and products. IRC was performed until the stationary point was reached with RMS gradient less than 1 × 10−4 hartree per bohr. Stationary points located through IRC were then completely optimized with the above mentioned method. The solvent effect was studied through single point energy calculations in methanol solvent through the polarization continuum model (PCM). The reported energies for all structures are Gibbs free energies (in kcal mol−1).Results and discussion
To explore possible reaction mechanisms for the formation of 3 and 2 from 1, we have performed DFT calculations at the B3PW91 level of theory. Theoretical studies on oxidative esterification and amidation with other late transition metals (Ru, Pd, and Au) reveal the occurrence of three possible mechanisms: inner sphere, outer sphere and intermediate sphere mechanisms.36–46 In the inner sphere mechanism, a coordinated alcohol delivers a β-hydride to the metal (Fig. 1). In the outer sphere mechanism,47 the alcohol delivers a hydride to the metal, but without being coordinated to the metal. Both inner sphere and outer sphere mechanisms involve the transfer of hydride to metal; however, in the intermediate sphere mechanism, hydrogen is transferred to a hydrogen acceptor instead of a metal. | ||
| Fig. 1 Catalytic cycle for metal [Ru] catalyzed dehydrogenative ester formation. | ||
The inner sphere mechanism involving the liberation of hydrogen is proposed in the oxidative esterification by the ruthenium pincer complex (Fig. 1). An inner sphere mechanism is also proposed for the palladium catalyzed esterification.48 However, a bifunctional double hydrogen transfer mechanism is shown to operate for the ruthenium dehydrogenation catalyst49 originally discovered by Milstein.13 Both inner and outer sphere mechanisms are shown to operate in the conversion of aliphatic amino alcohols to amino aldehydes.50 Inner and outer sphere mechanisms involve a hydride abstraction by metal (vide supra). β-Hydride elimination by metal requires a vacant coordination site and an electron in the d orbital of the metal. Considering these requirements, zinc species with more than three ligands are not anticipated to be active catalysts for β-hydride elimination. With these facts in mind, it would be interesting to explore the operating mechanism in zinc catalysed oxidation reactions. A general numbering scheme for discussion of compounds in this manuscript is given in Fig. 2.
 | ||
| Fig. 2 Numbering scheme for discussion of the computational results. | ||
The experimental reaction conditions show that the oxidative esterification reaction may be carried out in ZnBr2 alone, without any added ligand, but the yields are low. Addition of ligands 4–8 significantly improves the conversion yields. Efficiencies of ligand 4 and 7 are much better than 5, 6 and 8. Because the efficiencies of ligands 4 and 7 are comparable, ligand 7 is used for this study to reduce the computational cost. In the study by Wu, H2O2 is used as the oxidant. In a similar report on oxidative amide formation from alcohol and amine, ZnI2 and TBHP are used as the catalyst and oxidant, respectively. In this study, mechanistic details of a ZnBr2 catalyzed reaction in the presence of H2O2 oxidant are studied and the effects of ligand, oxidant and catalyst are also explored.
Initially, we studied the inner sphere mechanism for zinc catalysed oxidation of alcohol to ester (Fig. 3). The complex 9 is an eighteen electron complex and has a distorted tetrahedral geometry around the zinc atom. The complex 9 has another isomer 10, which is lower in energy than 9 by 19.01 kcal mol−1. A proton from carboxylic oxygen (O8) in 9 is shifted to O1 in complex 10. The complex 10 is occasionally obtained during the optimization of complex 9.
 | ||
| Fig. 3 Energy profile for oxidation of alcohol to aldehyde through β-hydride elimination (inner sphere mechanism) by zinc catalyst, calculated at B3PW91/6-311G(d,p) with SDDALL pseudo potential for Zn and Br. All energy values are relative to 9 at 0 kcal mol−1. All bond lengths are in Angstroms. Unnecessary hydrogen atoms are removed for clarity. | ||
A transition state TS9–11 for β-hydride elimination by zinc is located at a barrier of 31.78 kcal mol−1 from 9. This activation barrier is considerably higher than the one reported for the ruthenium based catalyst (8.80 kcal mol−1).49,50 The high activation barrier is not unexpected because the ability of transition metals to abstract β-hydride decreases from left to right in the periodic table; moreover, it is expected to be small for zinc. There are certain marked differences in the transition states for Ru and zinc based species. The transition state for β-hydride elimination to ruthenium catalyst is a very early transition state with geometry very similar to the starting material,50 whereas the zinc based transition state is almost in the middle. The geometry around zinc in the transition state is close to distorted square pyramid. The C2–H6 bond is considerably elongated to 1.73 Å in the transition state from 1.10 Å in 9. Moreover, the Zn–H6 and O–Zn bond distances are 1.69 Å and 2.13 Å, respectively. A few important structural parameters are given in Table 1. The geometry around zinc in 11 is distorted tetrahedron. The aldehyde product is no longer in coordination with the metal centre and shows hydrogen bonding interaction with the carboxylic acid moiety of the ligand. N7–Zn–H6, O1–Zn–O8 and N7–Zn–O8 bond angles are 116.24°, 113.35° and 72.15°, respectively. The hydride shift on zinc is also thermodynamically uphill by 4.04 kcal mol−1 from 9. The kinetic barrier for the hydride elimination is much higher than the similar reaction with other late transition metals (vide supra), which may be attributed the electron rich nature of the zinc metal, especially with four ligands coordinated. Because the reaction is carried out experimentally at room temperature, the kinetic barrier for β-hydride elimination is very high to be accessible under the reaction conditions. Therefore, β-hydride elimination (inner sphere) is not believed to be a plausible mechanism.
| Bond | 9 | TS9–11 | 11 |
|---|---|---|---|
| C–H | 1.10 | 1.73 | 5.11 |
| Zn–H | 3.08 | 1.69 | 1.54 |
| Zn–O | 1.84 | 2.13 | 4.16 |
| C–O | 1.40 | 1.27 | 1.22 |
The kinetic barrier of the inner sphere mechanism is very high to be accessible under the experimental reaction conditions; therefore, the intermediate sphere mechanism is envisaged. In the intermediate sphere mechanism, the hydrogen acceptor (H2O2 in this case) also coordinates to the metal. The complex 12 is a 20 electron species with a distorted square pyramid type structure. The alkoxide oxygen (O1) is strongly coordinated to zinc at the distance of 1.87 Å. An oxygen atom of H2O2 (O9) is weakly coordinated to zinc (2.30 Å), but this interaction is strong compared to the coordination of carboxylic oxygen with zinc (2.42 Å). The other OH (O10) proton of H2O2 interacts with the bromide ligand. Quite similar to complex 9, a proton shifted isomer of 12 is more stable than the complex 12. The complex 13 is structurally similar to 12 except that a proton from O8 is shifted to O1. The complex 13 is 20.06 kcal mol−1 more stable than 12 and this energy difference is very similar to the difference in energies of 9 and 10 (19.01 kcal mol−1).
A transition state for the hydrogen shift to hydrogen peroxide is located at a barrier of 22.63 kcal mol−1. The kinetic barrier is remarkably low and easily accessible under the reaction conditions than the kinetic barrier for the inner sphere mechanism (31.78 kcal mol−1). In the transition state, C2–H6 and O9–O10 bonds are elongated to 1.23 and 1.80 Å, respectively. Moreover, both oxygens of H2O2 are now coordinated to zinc (2.28 and 2.66 Å). This additional interaction with zinc is probably responsible for the low activation barrier of the intermediate sphere mechanism. An ortho hydrogen of the pyridine moiety also shows interaction with O10 of H2O2 (2.06 Å). The reaction is thermodynamically favourable by 78.78 kcal mol−1 from 12. The high energy of reactions may be due to the factors as follows: the weak O9–O10 bond in the starting complex 13 is replaced by the strong O–H bond, and the OH moiety generated during the reaction is also coordinated to the metal.
The product of the reaction (14) is a penta-coordinate complex. The water molecule produced (by the acceptance of hydrogen by H2O2) shows hydrogen bonding interactions with the OH ligand and the aldehyde fragment. The water molecule acts as a hydrogen bond acceptor and donor for aldehyde (2.13 Å) and OH (1.71 Å) ligands, respectively. The OH group of the carboxylic acid has very weak interaction with the metal and the bond is elongated to 2.42 Å (see Fig. 4 for details). The N7–Zn and O–Zn bond distances are 2.18 and 2.13 Å, respectively. The kinetic barrier predicted through this mechanism is in good agreement with the experiment, which suggests that the intermediate sphere mechanism may be a plausible one. Another experimental aspect in support of this mechanism is the acceleration of the reaction with higher concentration of H2O2 and it may be due to the direct involvement of H2O2 in the rate determining step (vide supra).
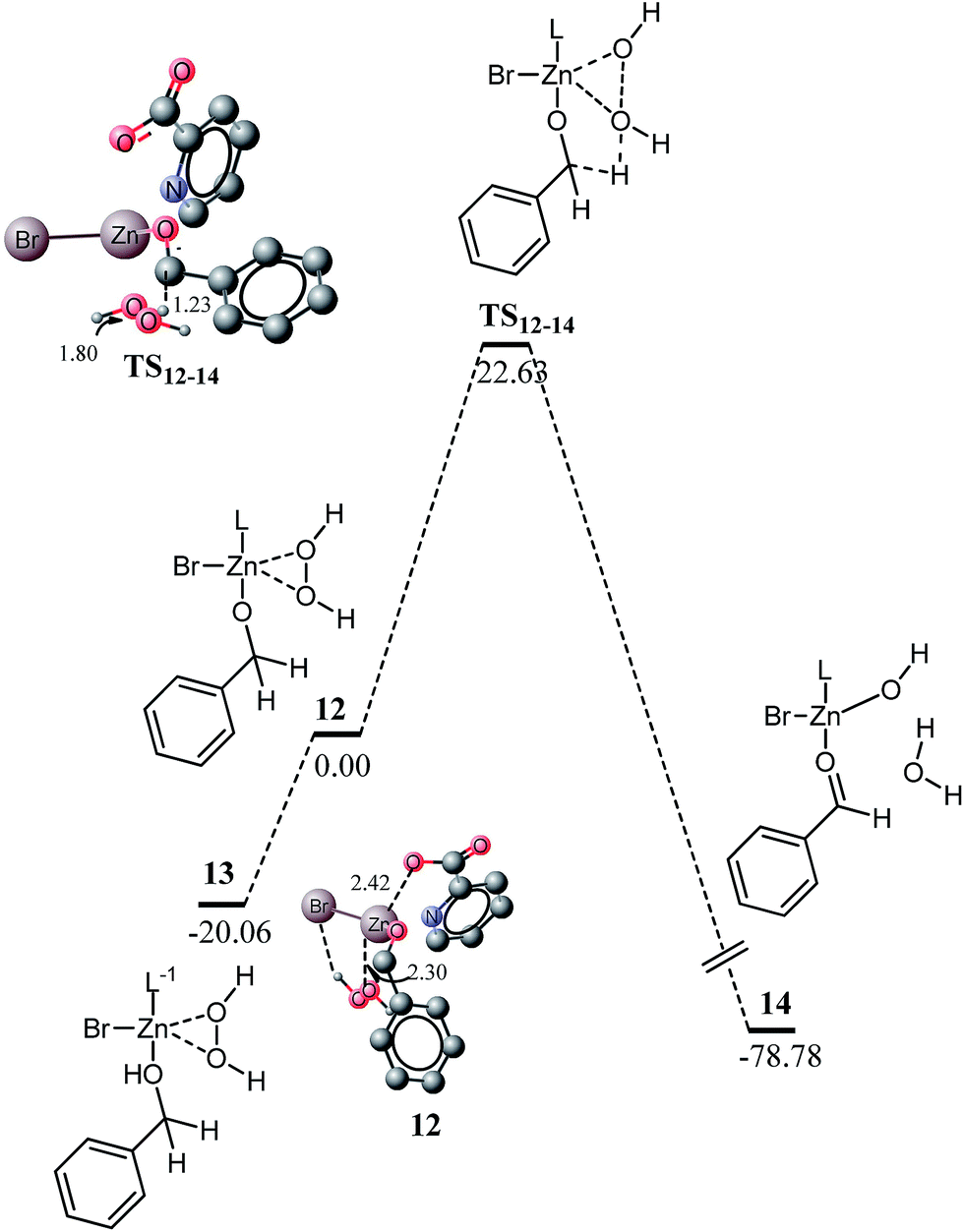 | ||
| Fig. 4 Energy profile for the oxidation of alcohol to aldehyde through H2O2 coordinated to a zinc catalyst, calculated at B3PW91/6-311G(d,p) with SDDALL pseudo potential for Zn and Br. All energy values are relative to 12 at 0 kcal mol−1. All bond lengths are in Angstroms. Unnecessary hydrogen atoms are removed for clarity. | ||
In similar reactions with other metals, an accepted notion is that the generated aldehyde reacts with alcohols to generate hemiacetals. The latter then enters into the catalytic cycle again and generates the ester product through another oxidation. Oxidation of aldehydes to esters is carried out with a variety of alcohols26,28 for structural diversity in esters; however, we have chosen butanol as an example. The formation of hemiacetal and its coordination to zinc by replacing water molecules is not analyzed in this study. In the intermediate sphere mechanism, the next step is the exchange of a proton with a hydroxyl ligand to form an alkoxy ligand. Although the proton exchange is not believed to be a kinetically demanding step, this reaction is modelled as well. The substrate may have many possible conformations and orientations of the aliphatic chain on oxygen with respect to the catalyst. A systematic search of all the possibilities in the intermediates and transition states was not carried out because they are not expected to significantly influence the energy profiles. The hemiacetal bound complex 15 has a tetrahedral geometry around zinc. The hemiacetal oxygens are not coordinated with zinc in 15, rather the hemiacetal is held there by hydrogen bonding interactions with other ligands (OH and L). The OH of the hemiacetal has hydrogen bonding interaction with the hydroxyl ligand on Zn (O–H⋯O bond distance is 1.56 Å). The O11 of the hemiacetal interacts with carboxylic oxygen (O10) at a distance of 1.61 Å. The Zn–Br distance in the complex is very similar to other complexes (2.46 Å). Zn–O (OH), Zn–N7 and Zn–O8 bond distances are 1.93, 2.16 and 2.20 Å, respectively.
All attempts to locate a transition state for the proton shift in 15 met with failure. It may be argued that the proton shift may be a barrierless process, at least at this level of theory. Alternatively, it may be argued that the transition state for this reaction resides on a shallow potential energy surface and soon relaxes to either reactant or product for the reaction. The proton shift is thermodynamically an uphill process by 5.35 kcal mol−1. Surprisingly, a transition state for the proton shift could be located at a barrier of 6.62 kcal mol−1 from 15 when a lower level of theory was applied (B3PW91/6-31G(d)) (for C, H, N and O) and LANL2DZ (Br and Zn). Most of the structural parameters remain constant on going from 15 to the transition state, TS15–16, with the exception of oxygen hydrogen bond distances at the active site.
Hemiacetal coordinated to zinc can be converted into an ester in a process very similar to the conversion of 12 to 14 (Fig. 4). A transition state for the intermediate sphere mechanism is modelled for this oxidation. Complex 17 is the active specie for this reaction, generated by replacement of a water molecule in 16 with hydrogen peroxide. A transition state for the oxidation of hemiacetal to ester is located at a barrier of 21.99 kcal mol−1. The structural features of the transition state are very similar to TS12–14. A few important structural features are given in Fig. 5. The reaction is highly exothermic by 90.15 kcal mol−1 and the exothermicity is even higher than that for 12 to 14.
 | ||
| Fig. 5 Energy profile for the oxidation of hemiacetal to ester through H2O2 coordinated to the zinc catalyst, calculated at B3PW91/6-311G(d,p) with SDDALL pseudo potential for Zn and Br. All energy values are relative to 17 at 0 kcal mol−1. Unnecessary hydrogen atoms are removed for clarity. | ||
Kinetic barriers for the intermediate sphere mechanisms (Fig. 4 and 5) are quite consistent with the experimental reaction conditions, which suggest that the intermediate sphere mechanism is a plausible mechanism for the conversion of alcohols into esters through zinc catalysis. The outer sphere mechanism is another possibility besides the inner and intermediate sphere mechanisms. However, the outer sphere mechanism was not investigated primarily for two reasons: (a) the outer sphere mechanism also involves a hydride shift to zinc, which is believed to be kinetically highly unfavourable (very similar to the inner sphere mechanism) because it involves a hydride shift to an electron rich zinc atom; (b) the outer sphere mechanism also requires a proton acceptor at a suitable position, which is missing in this catalytic system.
A number of zinc mediated oxidative transformations were recently reported in the literature, wherein variation in zinc halides (ZnI2, ZnBr2, and ZnCl2), oxidant (TBHP, H2O2) and ligands have been studied. Some of these reactions involve significant changes in the catalytic system. For example, ZnI2 and TBHP are used as the catalyst and oxidant, respectively, in the oxidative transformation of alcohol and amines to amides without any added ligand. Therefore, in this study, the effect of oxidant and zinc catalyst is studied and finally, the kinetics and thermodynamics of a ligandless reaction are also investigated. In the first approach, the effect of the oxidant is investigated, wherein H2O2 is replaced with TBHP and the potential energy diagram for the intermediate sphere mechanism is explored. The potential energy for the inner sphere mechanism is not affected with changes in the oxidant because the oxidant is not involved in the β-hydride elimination. The potential energy diagram for the oxidation of alcohol to ester with TBHP (through intermediate sphere mechanism) is very similar to the H2O2 mediated reaction. Replacement of a hydrogen atom with a tert-butyl group does not cause significant changes in the geometry of complex 20. The interaction of the hydroxyl proton O10 (of H2O2) with the bromide ligand causes some deformation of the geometry of the complex 12 (Fig. 6). The geometries and related structural parameters are shown in Fig. 6.
 | ||
| Fig. 6 Optimized geometries and related structural parameters for 12 and 20. | ||
Structure 21 is an isomer of compound 20, which lies about 6.81 kcal mol−1 lower in energy than 20 (Fig. 7). In 21, the benzyloxy moiety does not show hydrogen bonding interaction with the carboxylic acid part of the ligand, rather it shows non-bonding interactions with the OH of TBHP. The activation barrier for the oxidation to aldehyde through TBHP oxidant was calculated at 23.06 kcal mol−1. The activation barrier with TBHP is slightly higher than the corresponding reaction with H2O2 (22.63 kcal mol−1). Changing the oxidant from H2O2 to TBHP affects the interactions in the transition states as well. With H2O2, the hydroxyl (O10) proton shows non-bonding interactions with the bromide ligand, whereas with TBHP, a proton from the tertiary butyl group shows non-bonding interactions with bromide. Most of the other structural features are very similar for both transition states, except C2–H6 and Zn–OBu/OH bond distances. These distances are small with H2O2 and probably make the TS12–14 more stable than TS20–22. The reaction is highly exothermic, quite similar to the reaction with H2O2. The energy profile for proton shift in 23 to deliver 24 is very similar to what is observed for the transformation of 15 to 16. The transition state could not be located at a higher level of theory; however, it could be located at a lower level (Fig. S1 and S2†).
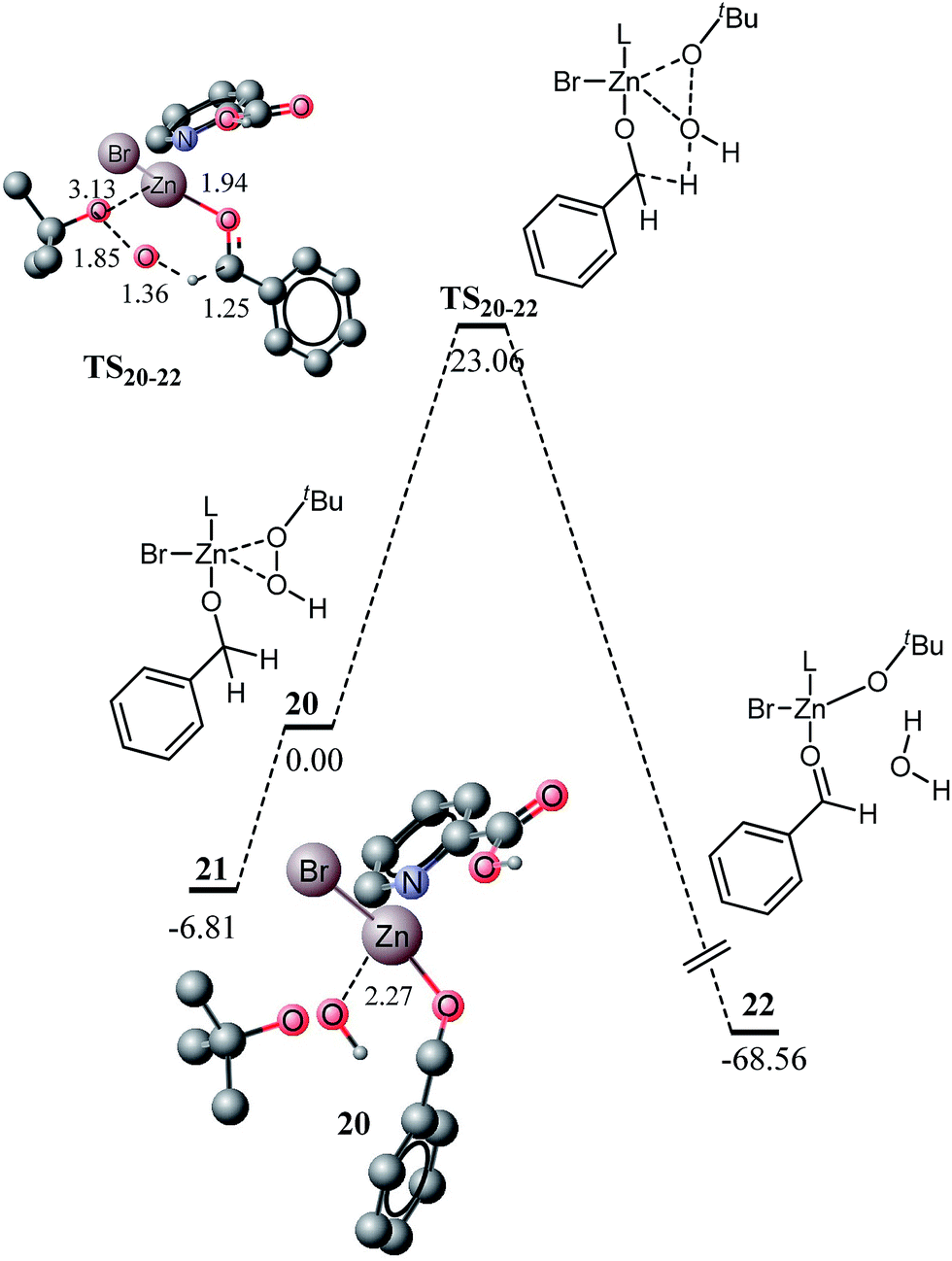 | ||
| Fig. 7 Energy profile for the oxidation of alcohol to aldehyde through TBHP coordinated to the zinc catalyst calculated at B3PW91/6-311G(d,p), with SDDALL pseudo potential for Zn and Br. All energy values are relative to 20 at 0 kcal mol−1. All bond lengths are in Angstroms. Unnecessary hydrogen atoms are removed for clarity. | ||
Hydrogen transfer from the hemiacetal to TBHP in 25 (to deliver the ester complex 26) (Fig. S3†) has a kinetic barrier of 18.75 kcal mol−1, which is lower than the oxidation of benzylic alcohol to benzaldehyde under TBHP conditions (TS20–22). This trend is consistent with what is observed with H2O2, i.e., activation barriers for the oxidation of hemiacetals to esters are lower than for the oxidation of benzylic alcohols to aldehydes; however, the activation barrier is much lower with TBHP. The kinetic barrier for the oxidation to esters with TBHP is lower than with H2O2 by about 3.24 kcal mol−1. A comparison of the energy profiles for reactions under TBHP and H2O2 conditions illustrate that H2O2 is a better oxidant in the intermediate sphere mechanism for the oxidation to aldehydes, whereas TBHP is superior for oxidation to esters. These theoretical findings are consistent with the experimental observations.33
To explore the effect of halide ligands on zinc, TBHP coordinated complexes were analysed. Hydrogen transfer from the zinc iodide coordinated benzylic alcohol 27 to generate benzaldehyde 28 (Fig. S4†) is kinetically less demanding by 4.70 kcal mol−1 than the similar reaction in 20. However, an increase in activation barrier for the ester formation step is observed (21.53 kcal mol−1). The transition state from 29 lies at a barrier of 21.53 kcal mol−1 compared to 18.75 kcal mol−1 from 25. These theoretical calculations are in accordance with the practical observations. Under similar conditions, 61% conversion has been observed experimentally with ZnBr2, whereas only 50% conversion is reached with ZnI2. Moreover, for ZnBr2, the ester is the dominant product, whereas ZnI2 delivers only the aldehyde product. The oxidation of aldehyde to ester using ZnI2 is hindered by the higher activation barrier for the second oxidations step (see Fig. S5†).
Finally, the effect of the ligand (pyridine-2-carboxylic acid) on the oxidative esterification was explored. Because the zinc complex without the pyridine-2-carboxylic acid ligand is a less than 18 electron species, it is interesting to investigate whether β-hydride elimination is a favourable process in these complexes or not. The activation barrier for the hydride shift is about 30.94 kcal mol−1 with respect to 30a. The activation barrier for the hydride shift is relatively decreased in the absence of the ligand (30.94 vs. 31.78 kcal mol−1) (see Fig. S6†). In the transition state (TS30a–31), C2–H and Zn–H bond lengths are 1.62 and 1.68 Å, respectively. Moreover, the reaction is thermodynamically uphill. The product of this step, 31, lies 6.23 kcal mol−1 higher in energy than 30a.
Because the activation barriers associated with the inner sphere mechanism (shown in Fig. S6†) are slightly decreased in the absence of ligand (although highly inaccessible at room temperature), it was of great interest to investigate the energetics of the ligandless intermediate sphere mechanism. A transition state was located for a hydrogen shift to TBHP with concomitant breakage of the O–O bond at barrier of 29.37 kcal mol−1 from 32a. In the transition state, C1–H8 and H8–O6 bond lengths are 1.21 and 1.52 Å, respectively. The O6–O7 bond of TBHP is considerably elongated (1.81 Å compared to 1.44 Å in 32a). The transition state is of relatively lower energy compared to the one for the hydride shift to zinc and the overall reaction is highly exothermic. The activation barrier for the hydrogen shift to TBHP in the absence of ligand is about 7 kcal mol−1 higher than with the ligand. This is again consistent with the experimental observation wherein 100% conversions were achieved in the presence of ligand compared to 50–60% conversion in the absence of ligand. Although the activation barrier for the intermediate sphere mechanism is increased in the absence of ligand, the activation barrier is lower than for the inner sphere mechanism (β-hydride elimination). These calculations reveal that the intermediate sphere mechanism is the plausible mechanism in the presence and absence of ligands. The water molecule generated as a result of the hydrogen shift also coordinates with the zinc atom, although this oxygen atom is quite a distance away from zinc in the starting material 32a (compare 2.30 Å in TS32a–33 with 2.91 Å in 32a). The low activation barrier may be attributed (Fig. S7†) to the formation of the metal oxygen bond in the transition state compared to the relatively unstable M–H bond in the TS30a–31.
Conclusions
In summary, the mechanism of the zinc(II) catalyzed oxidative transformation of alcohols to esters using H2O2 as oxidant was investigated by density functional theory methods, and the mechanistic outcomes were compared with those of other late transition metals. Both inner sphere and intermediate sphere mechanisms were analyzed in the presence and absence of pyridine-2-carboxylic acid (ligand). The inner sphere mechanism, which involves a hydride transfer to the transition metal, was found to be kinetically more demanding than the competitive intermediate sphere mechanism. In the presence of the ligand, the calculated activation barrier for β-hydride elimination (inner sphere) was 31.78 kcal mol−1. The calculated activation barriers for the hydrogen shift from the alkoxy ligand (intermediate sphere mechanism) to the oxidant (H2O2) were 22.63 and 21.99 kcal mol−1 for the aldehyde and ester formation steps, respectively. Ligands significantly affected the inner and intermediate sphere mechanisms. In the absence of ligand (pyridine-2-carboxylic acid), the activation barrier for β-hydride elimination increased slightly; however, the effect was more pronounced for the intermediate sphere mechanism wherein the activation barrier for the rate determining step decreased by more than 7 kcal mol−1. The effect of halide (on zinc) was relatively small. The presence of an iodide on zinc instead of a bromide provided easy access to aldehydes because of the lower activation barrier for the first step, whereas the activation energy for the second step was significantly increased. Moreover, steric bulk in the oxidant such as TBHP increased the activation barrier for the oxidation of alcohols to esters by 0.43 kcal mol−1; however, the activation barrier for the oxidation of hemiacetals to esters was decreased by 3.24 kcal mol−1. The mechanism of zinc catalyzed oxidative esterification is different from the mechanism of the same reaction with other late transition metals. With Ru, Pd and Ir, inner or outer sphere mechanisms carry out the reactions. The intermediate sphere mechanism for zinc is an interesting aspect for further exploration.Acknowledgements
The authors acknowledge the financial support from Higher Education Commission of Pakistan (Grant No. 1899, 2469, 2981), COMSATS Institute of Information Technology, and University of Rostock. This study has been partly supported by the BMBF within the project “Light2Hydrogen” (Spitzenforschung und Innovation in den Neuen Landern), by the European Union (European Social Funds, ESF) within the project “PS4H” and by the Ministry for Education, Science and Culture of Mecklenburg-Vorpommern.Notes and references
- J. Otera, Esterification: Methods, Reactions, and Applications, WileyVCH, Weinheim, 2003 Search PubMed.
- Modern Polyester: Chemistry and Technology of Polyester and Copolyesters, ed. J. Scheirs and T. E. Long, WileyVCH, 2003 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, USA, 2nd edn, 1999 Search PubMed.
- H. Finkelstein, Ber. Dtsch. Chem. Ges., 1910, 43, 1528–1532 CrossRef CAS.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed.
- Compendium of Organic Synthetic Methods, ed. I. T. Harrison and S. Harrison, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1971, vol. 1 Search PubMed.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed. Engl., 2009, 48, 4114–4133 CrossRef PubMed.
- S. K. Rout, S. Guin, K. K. Ghara, A. Banerjee and B. K. Patel, Org. Lett., 2012, 14, 3982–3985 CrossRef CAS PubMed.
- X.-F. Wu and C. Darcel, Eur. J. Org. Chem., 2009, 2009, 1144–1147 CrossRef.
- S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed. Engl., 2011, 50, 5139–5143 CrossRef CAS PubMed.
- C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed. Engl., 2011, 50, 5144–5148 CrossRef CAS PubMed.
- M. C. R. Guerbet, Acad. Sci., Paris, 1899, 128, 1002 Search PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- X.-F. Bai, F. Ye, L.-S. Zheng, G.-Q. Lai, C.-G. Xia and L.-W. Xu, Chem. Commun., 2012, 48, 8592–8594 RSC.
- C. Liu, S. Tang and A. Lei, Chem. Commun., 2013, 49, 1324–1326 RSC.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366 RSC.
- H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776 RSC.
- C. Gunanathan, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2009, 131, 3146–3147 CrossRef CAS PubMed.
- N. Yamamoto, Y. Obora and Y. Ishii, J. Org. Chem., 2011, 76, 2937–2941 CrossRef CAS PubMed.
- F.-Z. Su, J. Ni, H. Sun, Y. Cao, H.-Y. He and K.-N. Fan, Chemistry, 2008, 14, 7131–7135 CrossRef CAS PubMed.
- M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed. Engl., 2012, 51, 5711–5713 CrossRef CAS PubMed.
- J. Zhao, C. Mück-Lichtenfeld and A. Studer, Adv. Synth. Catal., 2013, 355, 1098–1106 CrossRef CAS.
- L. Wang, J. Li, W. Dai, Y. Lv, Y. Zhang and S. Gao, Green Chem., 2014, 16, 2509–2514 RSC.
- Y. Zhu and Y. Wei, Eur. J. Org. Chem., 2013, 2013, 4503–4508 CrossRef CAS.
- X.-F. Wu, Chemistry, 2012, 18, 8912–8915 CrossRef CAS PubMed.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- X.-F. Wu, Tetrahedron Lett., 2012, 53, 3397–3399 CrossRef CAS.
- Z.-Z. Song, J.-L. Gong, M. Zhang and X.-F. Wu, Asian J. Org. Chem., 2012, 1, 214–217 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Rev. C.01, Wallingford CT, 2009 Search PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. H. Dunning and P. H. Hay, Modern Theoretical Chemistry, ed. H. F. Schaefer, New York, USA, 1976 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. P. Perdew, Electronic structure of Solids, ed. P. Ziesche and H. Eschrig, Akademic Verlag, Berlin, 1991 Search PubMed; K. Burke, J. P. Perdew and Y. Wang, in Electronic Density Functional Theory: Recent Progress and New Directions, ed. J. F. Dobson, G. Vignale and M. P. Das, Plenum, New York, 1998, p.81 Search PubMed.
- A. Nova, D. Balcells, N. D. Schley, G. E. Dobereiner, R. H. Crabtree and O. Eisenstein, Organometallics, 2010, 29, 6548–6558 CrossRef CAS.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051–1069 CrossRef CAS.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- R. Noyori, Asymmetric catalysis: Science and opportunities (nobel lecture), Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS PubMed.
- R. M. Bullock, Chem.–Eur. J., 2004, 10, 2366–2374 CrossRef CAS PubMed.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- T. Ikariya, Top. Organomet. Chem., 2011, 37, 31–53 CrossRef CAS.
- T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC.
- O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21–27 RSC.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- A. Comas-Vives, G. Ujaque and A. Lledós, Inner- and Outer-Sphere Hydrogenation Mechanisms: A Computational Perspective, 2010, vol. 62 Search PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS PubMed.
- W. Hu, J. Li, S. Deng, J. Huang, X. Le and W. Zheng, J. Organomet. Chem., 2013, 740, 10–16 CrossRef CAS.
- D. Cho, K. C. Ko and J. Y. Lee, Organometallics, 2013, 32, 4571–4576 CrossRef CAS.
- A. Nova, D. Balcells, N. D. Schley, G. E. Dobereiner, R. H. Crabtree and O. Eisenstein, Organometallics, 2010, 29, 6548–6558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Total electronic, zero-point and Gibbs free energies along with the Cartesian coordinates of the optimized geometries. See DOI: 10.1039/c5ra23096a |
| This journal is © The Royal Society of Chemistry 2016 |