
Noninvasive target CT detection and anti-inflammation of MRSA pneumonia with theranostic silver loaded mesoporous silica
Hao Zhanga,
Qingqing Dinga and
Jing Ding*b
aDepartment of Geriatric Gastroenterology, The First Affiliated Hospital with Nanjing Medical University, Nanjing, People's Republic of China
bDepartment of Respiratory Medicine, the Affiliated Nanjing Children Hospital with Nanjing Medical University, Nanjing, 210008, People's Republic of China. E-mail: djnjch@sina.com; Fax: +86-25-83304239; Tel: +86-25-83117319
First published on 5th January 2016
Abstract
In this study, the non-invasive diagnosis of methicillin resistant Staphylococcus aureus (MRSA) related pneumonia via X-ray CT and related anti-inflammatory profiles post theranostic silver loaded mesoporous silica nanomedicine is reported. Mesoporous silica NPs were firstly synthesized and chemically modified to covalently bond to silver clusters and anti-MRSA antibodies. The structure of the hybrid was investigated via transmission electron microscopy, dynamic light scattering analysis and X-ray photoelectron spectra. MTT and broth dilution assays confirmed the biocompatibility and antibacterial potency of the platform. In vivo CT study of rats bearing MRSA pneumonia and anti-inflammatory profiles as bronchoalveolar lavage and pathological assays give evidence to the theranostic potential of our platform. Biochemical and hematological assays confirmed the long term safety of the nanomedicine for in vivo application.
Introduction
The prevalence of antibiotics resistant pathogen related infection has raised global concern for healthcare.1–3 Among them, the methicillin-resistant Staphylococcus aureus (MRSA) pathogen, which secretes highly cytotoxic virulence factors (e.g. Panton-valentine leukocidin), has been proved to contribute primarily to hospital related infection according to Rubinstein.4 Considering the infection related mortality, hospital acquired pneumonia especially ventilator-related pneumonia, containing staphylococcal cassette chromosome (SCC) type I–III, has been considered to feature a poor prognosis.5,6 When the infected host is ICU related, mortality rates even higher than 70% could be seen 48 h post infection.7 Thus, finding a suitable assay for early characterization and treatment of MRSA pneumonia is of significant practical importance. Traditional bacteriological assays like blood culture failed to provide adequate efficacy and sensitivity for routine clinical practice.8 Other invasive methods like bronchoalveolar lavage fluid (BALF) culture or protected specimen brush also suffer from limited specificity, which makes it impossible to track the therapeutic outcome.9 Much effort has been devoted to the development of a feasible and reliable non-invasive way to diagnose MRSA related pneumonia. At present, Da has reported the successful detection of MRSA pneumonia via computed tomography (CT) imaging and related anti-inflammation profiles by taking advantage of theranostic MRSA targeting Au@Ag NPs.10 Though these findings are encouraging, the high cost of gold remarkably hindered the practical translation of the Au@Ag NPs. Herein, we report a novel cost-effective therapeutic platform for the same purpose. Mesoporous silica NPs were firstly synthesized according to Shi et al. and modified to load silver clusters integrated with both X-ray attenuation and anti-inflammatory potency.11 The hybrid NPs were further anchored with polyethyleneglycol (PEG) molecules and anti-MRSA antibodies to render the platform solubility and targeting capability, respectively. In vitro and in vivo tests collectively confirmed that effectiveness of our platform. Long term cytotoxicity study provided strong evidence to the safety and biocompatibility of our platform. Therefore, we hope our therapeutic nanomedicine could play an important role in future clinical trials.Materials and methods
Materials
Cetyltrimethylammonium bromide (CTAB), tetraethoxysilane (TEOS) and sodium hydrate (NaOH) were purchased from Nanjing Chemical Reagent Co. Ltd. (3-Aminopropyl) triethoxysilane (APTES), meso-2,3-dimercaptosuccinic acid (DMSA), silver nitrate, ethyl dimethylaminopropylcarbodiimide (EDC), sulfo-NHS and 4-(N-maleimidomethyl) cyclohexane-1-carboxylic acid 3-sulfo-N-hydroxysuccinimide ester sodium salt (SMCC) were purchased from Sigma-Aldrich. Phosphate buffer saline (PBS, pH = 7.4) and 4% paraformaldehyde were purchased from Hyclone (Thermo Scientific). mPEG-COOH (Mw = 5 KDa) was purchased from Keygen Co. Ltd. All the reagents mentioned herein were of analytical grade and used as received without further purification. Anti-MRSA antibody was purchased from Abcam (ab73263) and purified according to the manufacture instructions.Cells and pathogens
A549 (human alveolar basal epithelial cell line) was purchased from Type Culture Collection of Chinese Academy of Science (Shanghai, China). HPMES (human lung epithelial cell line), HRGEC (human kidney epithelial cell line) and HHSC (human hepatocellular cell line) were purchased from American Type Culture Collection (ATCC). DMEM medium (high glucose), fetal bovine serum (FBS) were purchased from Gibco (Life technology, America). Antibiotics as penicillin and streptomycin were purchased from Keygentec (Nanjing, China).E. coli (ATCC 11303), S. aureus (ATCC 27217), B. subtilis (ATCC 33712) and P. aeruginosa (ATCC 27853) were purchased from ATCC. While MRSA strain (SCC mec type II) was a kind gift from Department of Quanrantine in Nanjing Children's Hospital from a female host suffered from MRSA pneumonia.
Animal model
All the animal tests (including both normal rat and the disease bearing rat) were proved by the institutional ethical committee for animal care in Nanjing Medical University, and also were in accordance with the policy and protocols of the National Ministry of Health. Pathogens were firstly inoculated onto Columbia Blood Agar Base to recover susceptive faculties, and then the agar was changed every 24 hours to retain the activity of pathogens. Then, pathogens were harvested and re-suspended in 2 mL sterile PBS suspension to a final concentration of 106 CFU mL−1, kept the solution at 4 °C before applications. Ventilator-associated pneumonia model was established as previously described with a little modification.12 Briefly, male Wistar rats (4–6 week, average weight 180 g) were subjected to fast 12 h before operation and anesthetized via intraperitoneal urethane administration. Then an endotracheal tube with 16 gauge needle front was inserted into the trachea. After the placement of catheter, stethoscope was used to confirm the catheter was right in the trachea. Therefore, 80 μL freshly prepared PBS containing 106 CFU mL−1 pathogens was injected slowly to induce pneumonia. During the procedure, the rats were ventilated with a constant-volume respirator, and following parameters were adopted: inspired O2 fraction of 1.0; peak airway pressures of 8–12 cm H2O and a 2 cm positive end expiratory pressure (PEEP); as high as 15 mL kg−1 tidal volume was recommended every 30 minutes to avoid potential positional pulmonary atelectasis; breathing frequency 70–80 times per min; the respiratory rate was adjusted to maintain the pressure of CO2 between 35 and 50 mmHg. After the treatment, rats were left to recover on heated pad to avoid loss of heat.Characterization
The morphology of the as-obtained NPs were investigated with transmission electron microscope (TEM, JEOL TEM-100). Images of single particle were acquired with high resolution TEM (JEOL, TEM-2100). Hydration diameter and zeta potential properties were verified with Zetaplus instrument (Brookhaven Instruments Corporation, USA). For dynamic light scattering (DLS) analysis, all samples were filtrated through 0.22 μm cellulose filter before measurements. The scattered light of a vertically polarized He–Ne laser was collected by auto correlator at an angle of 90. For zeta potential analysis, each sample was adjusted to a concentration of 0.1% (w/v) in deionized water before measurement. All measurements were carried triplicate and final values were given in average. Surface element composition was investigated by X-ray photoelectron spectroscope (XPS, Thermo Fisher K-Alfa) with a focused monochromatic Al X-ray (1486.6 eV) source.Preparation of SLS NPs
The mesoporous silica NPs were synthesized via the soft template method according to Shi et al. reported.11 In brief, NaOH and CTAB solution were mixed together and vigorously stirred before the mixture become clear. Then, TEOS was added dropwise and kept reacting under magnetic stirring. After that, white precipitation was harvested via centrifugation and washed several times with PBS to remove unreacted TEOS. In order to remove the surfactant, the as-synthesized powder was re-dispersed in suspension containing ethanol and HCl and refluxed to remove the soft template (CTAB). Then, the NPs were collected via centrifugation and washed several times with ethanol and dried in vacuum.Surface modification was achieved by adding 50 mg freshly fabricated mesoporous Si NPs into 20 mL suspension (ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DI water 2:1/v/v) containing 30 mg APTES. The reaction was carried on for 48 h under reflux at 80 °C. Then the as-synthesized APTES functionalized mesoporous Si NPs were harvested via centrifugation and washed three times with ethanol to remove excess APTES and dried in vacuum. 120 mg EDC together with 87 mg sulfo-NHS were added into suspension containing 15 mg mPEG-COOH and 42 mg DMSA to activate the –COOH groups. After 3 h reaction, 36 mg above mentioned APTES modified Si NPs were added in and allowed to react for another 12 h at room temperature. After the PEGylation and DMSA conjugation, products were collected via centrifugation and subjected to several washing with ethanol and re-suspended in 10 mL DI water. Fresh prepared AgNO3 solution (0.1 M, 560 μL) was added and allowed to react for 80 min under magnetic stirring. Final product was isolated via centrifugation and purified with DI water washing to remove excess silver ion. Antibody conjugation was adapted as previously reported.13 Then, 3.2 mL freshly prepared SMCC modified anti-MRSA antibody solution (approximate 51.2 μg antibody) was added into the suspension. The conjugation process was carried out at 4 °C overnight. Excess antibody was removed via centrifugation and the products were purified with PBS washing and stored at 4 °C before further use (Scheme 1).
:DI water 2:1/v/v) containing 30 mg APTES. The reaction was carried on for 48 h under reflux at 80 °C. Then the as-synthesized APTES functionalized mesoporous Si NPs were harvested via centrifugation and washed three times with ethanol to remove excess APTES and dried in vacuum. 120 mg EDC together with 87 mg sulfo-NHS were added into suspension containing 15 mg mPEG-COOH and 42 mg DMSA to activate the –COOH groups. After 3 h reaction, 36 mg above mentioned APTES modified Si NPs were added in and allowed to react for another 12 h at room temperature. After the PEGylation and DMSA conjugation, products were collected via centrifugation and subjected to several washing with ethanol and re-suspended in 10 mL DI water. Fresh prepared AgNO3 solution (0.1 M, 560 μL) was added and allowed to react for 80 min under magnetic stirring. Final product was isolated via centrifugation and purified with DI water washing to remove excess silver ion. Antibody conjugation was adapted as previously reported.13 Then, 3.2 mL freshly prepared SMCC modified anti-MRSA antibody solution (approximate 51.2 μg antibody) was added into the suspension. The conjugation process was carried out at 4 °C overnight. Excess antibody was removed via centrifugation and the products were purified with PBS washing and stored at 4 °C before further use (Scheme 1).
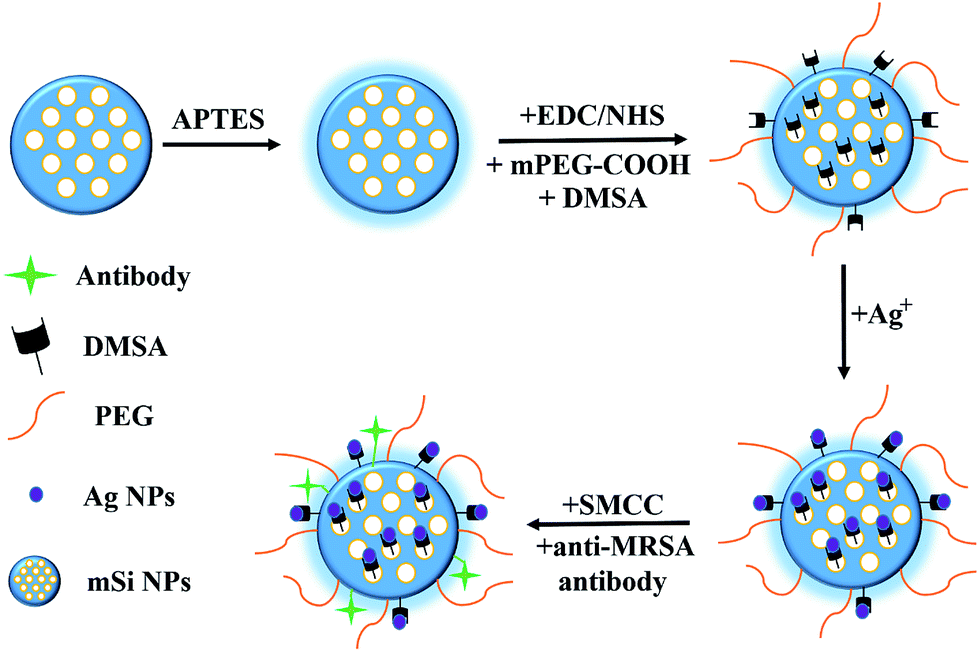 | ||
| Scheme 1 Synthesis of SLS NPs. | ||
Selectivity and antibacterial profiles
The binding efficacy of different platforms to pathogens (MRSA, S. aureus, E. coli, B. subtilis, P. aeruginosa) were assessed via well-established broth dilution assay. Briefly, sterile Eppendorf vials containing 2 mL PBS with 106 CFU mL−1 pathogens were prepared. Then, suspension containing 14 mg targeting or non-targeting SLS NPs were added into the above suspension separately. After 4 h incubation at 37 °C, pathogens were collected via centrifugation (2000 rpm × 10 min) and subjected to three washes with PBS. For the quantitative analysis of targeting efficacy, pathogens were dissolved in 2 mL aqua regia. After 24 h, supernatant of each tube was collected and quantity of silver element was determined by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500ce). To investigate the antibacterial capability of targeting and non-targeting SLS NPs, purified pathogens post incubation with NPs were re-suspended in broth medium and incubated at 37 °C for 48 h. Optical density (OD) at 570 nm of corresponding tube in different time intervals was measured (Biotek ELX800, USA). Pathogens suspensions free of antibacterial treatment were taken as control groups.The inhibition ratio (IR) was calculated as following:
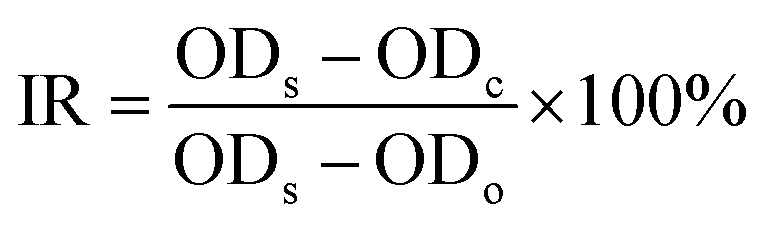
Cytotoxicity profile
Cytotoxicity of PEGylated and bare SLS NPs was tested via a flow-cytometry assay. A549, HPMES, HRGEC, HHSC cells were seeded in 6-well plates at a density of 5 × 105 cells per well in 4 mL of Iscoves modified Dulbeccos medium and incubated overnight at 37 °C in 5% CO2 atmosphere. The medium of each well was replaced with 2 mL fresh medium containing various concentrations of the NPs. All concentrations were tested in triplicates. After 24 h incubation, the medium was aspirated, and the cells were harvested with 0.25% trypsin (Sigma) treatment and washed twice with PBS. After that, cells were re-suspended in 200 μL binding buffer (BD Biosciences) followed by staining with Annexin V-FITC and propidium iodine (PI) solution. Flow cytometry analysis was performed on FACS verse (BD Biosciences) and analyzed using the Flowjo software (version 7.6.2, Tree star).CT imaging
For the in vivo imaging of pneumonia bearing rats, imaging modality and parameters were in consistent with in vitro imaging. Rats were firstly anesthetized with an intraperitoneal injection of 20 mg kg−1 ketamine and 10 mg kg−1 xylazine. After the anesthesia, suspensions containing Omnipaque®, targeting and non-targeting SLS NPs were administrated systematically for lung specific imaging. One bed position was scanned for 5 seconds to acquire the entire chest of the animal. CT images were collected in transversal view using liver window (L: 150 HU, W: 30 HU). Regions of interest, such as ground-glass opacity, bleeding, necrotic lesions, were analyzed by experienced doctors with the GE workstation.In vivo distribution
Both normal rats and ventilator associated pneumonia suffering rats were taken into consideration. 120 μL suspension containing 12 mg targeting and non-targeting SLS NPs was administrated systematically. Rats post treatment were sacrificed at different time intervals (n = 3). Lungs were harvested and dissolved in 2 mL aqua regia. The mixture was heated to boiling repeatedly until the solution became pellucid, replenished the mixture with DI water to reach a final volume of 1 mL. Silver concentration was determined with ICP-MS, and distribution profiles were presented as percentage of injected dose per gram organ as a function of time. Data was shown as mean ± SD.Anti-inflammatory effect
Rats subjected to different treatment were randomly picked from the group and sacrificed to take the necropsy and pathological analysis. Lungs were harvested and immobilized in 4% paraformaldehyde at 4 °C for 12 h, and then embedded into paraffin. Sections were further stained with hematoxylin and eosin (H&E). Observation was done by experienced physician under a light microscope at 100× magnification, and representative images were shown.Bronchoalveolar lavage (BAL) assay was taken to estimate the situation of inflammatory response in lung lesions. Rats were sacrificed and exsanguinated via the femoral vessel under sterile condition. After the thorax was opened, a 18 gauge needle was introduced to the trachea at the cricothyroid membrane. Then 1 mL sterilized PBS was administrated in and extracted out for several cycles to promote the exchange process between suspension and extracellular microenvironment in pneumonia sections. Upon treatment, IL-6 and TNF-α cytokine levels were characterized via enzyme linked immunosorbent assay (Elisa, R&D) according to manufacturer's instructions. Each analysis was carried out in triplicate and averaged results of statistical significance were given (P < 0.01).
Hematology and biochemical assays
Normal rats were sacrificed 1, 2 and 4 week post systematical injection of targeting SLS NPs (n = 3). Blood was separated into two parts, one was collected in sodium EDTA anticoagulant tube to analysis the levels of red blood cell (RBC), white blood cell (WBC), lymphocytes (LYM) and platelets (PLT). Another part was stored in gel containing coagulation tube to collect the serum. Levels of aminotransferase (ALT) and blood urea nitrogen (BUN) were verified.Results and discussion
Morphology of the as-synthesized silver cluster-loaded mesoporous silica nanoparticles (SLS NPs) was verified with TEM. Mono-dispersive spherical NPs with a mean diameter of 68.5 nm could be observed in Fig. 1. Dot like ultrafine NPs decorated on the mesoporous silica NPs exhibited much higher contrast with respect to Si NPs, demonstrate the successful loading of silver clusters and further confirmed by XPS (Fig. 1b). After surface PEGylation and antibodies conjugation, size distribution and surface zeta potential were also tested. As shown in Fig. 1c, surface modification with PEG and anti-MRSA antibody increased the hydration radius of SLS NPs from 79 ± 8 nm to 94 ± 11 nm. While the zeta potential changed dramatically from −21.44 eV to approximate neutral due to the PEGylation, in consistent previously reported, which is essential for further in vivo application to reduce non-specific opsonization.14 | ||
| Fig. 1 (A) TEM images of PEGylated SLS NPs, (B) XPS result of the SLS NPs and silver element, (C) DLS and zeta potential profiles of the SLS NPs pre and post PEGylation. | ||
Firstly, selectivity of targeting SLS NPs was firstly tested with respect to non-specific SLS NPs. After co-incubation of MRSA targeting SLS NPs and non-targeting SLS NPs with MRSA pathogens (105 colony forming unit per mL) with equivalent amount of SLS NPs (370 μg silver in 5 mg mesoporous Si NPs). From the SEM images (Fig. 2A), it could be clearly seen that targeting SLS NPs absorbed onto the out-membrane of MRSA bacteria without losing their initial structure or forming aggregations (inset Fig. 2A for targeting SLS NPs). To quantitatively determine the specificity, amount of absorption was investigated via ICP as shown in Fig. 2B. Target platform exhibited 8.735 fold higher binding efficacy to MRSA with respect to non-specific control. Both targeting and non-targeting platforms exhibited comparable binding capability to E. coli, S. aureus, P. aeruginosa regardless of the type of bacteria. Taken together, these findings suggested that surface modification with antibodies only rendered the platform with MRSA selectivity, in consistent with previously reported, which is essential for accurate targeting diagnosis.
 | ||
| Fig. 2 (A) SEM imaged of MRSA pathogens post incubation with targeting and non-targeting SLS NPs (inset: highlighted area on the membrane of single MRSA pathogen with red circle, white arrows pointed to SLS NPs). (B) Binding efficacy of targeting and non-targeting SLS NPs to MRSA, S. aureus, E. coli, B. subtilis, P. aeruginosa as determined by ICP-MS (**P < 0.001). | ||
Motivated by the superior targeting potency of our platform to MRSA pathogens, we were interested in their potential anti-bacterial capability. In Fig. 3, bacterias were incubated with medium containing targeting, non-targeting SLS NPs or vesicles and profiles of bacterial proliferation inhibition were recorded. For MRSA (Fig. 3A), targeting SLS NPs incubation leaded to pronounced reduction of bacterial viability (23% of control group 48 h post incubation) in contrast to non-targeting SLS NPs (87%) and vesicles (97%) without recurrence. In addition, remarkable loss of membrane structure of MRSA pathogens (Fig. 3B) give further solid evidences to the success of bactericidal effect rendered by targeting SLS NPs. For S. aureus and E. coli, both targeting and non-targeting platforms exhibit equivalent antibacterial capability over time and both higher than that of mSi NPs, but the differences were not of statistical significance. As we have already confirmed, targeting SLS NPs integrated higher binding efficacy only to MRSA pathogens, we speculated that selectivity of targeting SLS NPs to MRSA pathogens greatly increased the valid silver concentration close to MRSA bacteria and enhanced the anti-bacterial potency. Further studies might be needed to verify this envision.
 | ||
| Fig. 3 (A) Antibacterial profiles of targeting and non-targeting SLS NPs towards MRSA, S. aureus and E. coli as a function of incubation time via liquid broth dilution assay. (B) SEM images of MRSA pathogens post targeting SLS NPs treatment in low and high resolution (**P < 0.001; *P < 0.01). | ||
Cytotoxicity plays a pivotal role in determining the fate of nanomedicine during clinical translation. Before further in vivo application, we firstly tested the biocompatibility of the SLS NPs via flow-cytometry assay. As shown in Fig. 4, Annexin-V and PI were used to label early apoptosis related membrane phosphatidylserine and dead cells. Only Annexin-V positive stained cells were taken as early apoptotic with respect to Annexin-V and PI dual positive cells (late apoptotic). Taking the human lung adenocarcinoma epithelial cell line (A549) as models (Fig. 4A), SLS NPs without PEGylation exerted obvious cytotoxicity even in the presence of only 20 μg mL−1 silver (viability 89.4%; early apoptosis 9.64%; late apoptosis 0.92%), suggested the high cytotoxicity of bare silver clusters. Further increasing the silver concentration leaded to enhancement of apoptotic cells in both early and late stages, ruled out the potential in vivo application of SLS NPs absence of PEGylation. However, for SLS NPs with PEG modification, almost negligible loss of viability (<5%) could be seen post 24 h incubation with NPs containing less than 120 μg mL−1 silver element. Even when the concentration of silver was further elevated to 250 μg mL−1, only reasonable cytotoxicity for A549 could be observed (viability: 91.5%; early apoptosis: 1.9%; late apoptosis: 4.07%). We safely contributed the enhanced cell-tolerance to the protective role PEG coverage played. After that, we further tested the potential cytotoxicity SLS (with PEGylation without specific demonstration) exerted on normal epithelial cell lines from three major organs (Fig. 4B, HPMES, HRGEC and HHSC for lung, kidney and liver, respectively). It could be clearly seen that incubation with SLS NPs (120 μg silver mL−1) leaded to negligible loss of viability and further confirmed the biocompatibility of SLS NPs for in vivo application.
 | ||
| Fig. 4 Flow cytometry results of A549 cells post treatments with PEGylated or bared SLS in different concentration (A) and HPMES, HHSC, HRGEC cells co-cultured with PEGylated SLS NPs (120 μg silver mL−1) (C). (B and D) represented detailed fractions of cells in necrotic, late apoptotic, early apoptotic stage for (A) and (C), respectively. | ||
Iodine based molecules have been routinely used as CT contrast agents due to their superior X-ray attenuation capability and bio-compatibility. However, iodine based contrast agents suffered from several drawbacks including limited blood circulation half-life and non-specificity.15,16 In this study, to verify if our platform could exhibit superior target imaging potency, we tested in vivo performance of our platform in MRSA induced pneumonia with respect to both non-targeting SLS NPs and commercial available Omnipaque® contrast agent. As shown in Fig. 5A, it could be clearly seen that the target SLS NPs leaded to significant enhancement in the inflammatory lung 5 h post administration. As indicated by the 2D images from different layer (green dotted line), representative pneumonia imaging features like ground-glass opacity (white arrows) and caseous necrosis lesions (green arrows) caused by inflammatory cells, could be easily distinguished from lung parenchyma. HU profiles of randomly picked areas pre and post contrast further confirmed these results (Fig. 5B), HU values of all these sections elevated indicated that the inflammation response caused by pneumonia affected nearly the whole lung to different extent, which is hard to verify with traditional assays. In future works, our efforts will be given to unveil more pathological details with the aid of spectral CT during anti-inflammatory treatment in non-invasive manner. In contrast, no observable contrast enhancement could be seen post non-targeting SLS NPs or Omnipaque® injection, suggested that specificity of targeting SLS NPs enhanced the accumulation of SLS NPs in lung lesions. In addition, we investigated the in vivo distribution profile of both target and non-target SLS NPs in both normal and pneumonia bearing rats. As could be seen in Fig. 5C, accumulation of targeting SLS NPs in lung of normal rat was low (4.77%, 2.38% and 2.79% ID/g lung 12 h, 48 h and 96 h post administration, respectively). However, in the presence of MRSA pneumonia, lung accumulation of targeting SLS NPs obviously enhanced (15.47%, 25.39% and 22.95% ID/g lung 12 h, 48 h and 96 h post administration, respectively), significantly higher than that of non-targeting SLS NPs (5.87%, 3.75% and 2.26% ID/g lung 12 h, 48 h and 96 h post administration, respectively), demonstrated that the enhanced accumulation could be ascribed to active targeting rather than inflammation induced passive retention. Taken together, these findings strongly supported the role of MRSA antibody played in enhancing the target accumulation of SLS NPs in lung and contrast of MRSA related pneumonia lesions.
 | ||
| Fig. 5 (A) CT images of pneumonia bearing rats post contrast in transversal view post systematic Omnipaque®, targeting and non-targeting SLS NPs administration (white and green arrows indicate ground-glass opacity and caseous necrosis lesions, respectively). (B) Corresponding HU values of five randomly picked ROI in targeting SLS NPs group pre and post contrast. (C) In vivo bio-distribution profiles of targeting and non-targeting SLS NPs in lungs of both pneumonia bearing and healthy rats (**P < 0.001). | ||
Another important prospect of the study is the anti-inflammatory effect rendered by different platforms. Considering the high mortality rate of MRSA pneumonia within 48 h post infection, immediate inhibition of the MRSA pathogen proliferation and related inflammation response is of great importance in clinical trials. Therefore, we verified the therapeutic outcome of different platforms via pathological and molecular biological assays. As shown in Fig. 6A, inflammatory cytokines concluding TNF-α (pro-inflammatory cytokine related to tissue damage and acute inflammatory reactions)17 and IL-6 (T cell growth factor in amplifying lymphocyte response against foreign invasion)18 in BALF were quantitatively determined via Elisa assay. For targeting VI-mSi NPs treatment, levels of both cytokines decreased over time (TNF-α: from 66.9 pg mL−1 to 28.5 pg mL−1; IL-6: 430.4 pg mL−1 to 145.5 pg mL−1), strongly suggested the reduced inflammatory response. In contrast, fluctuation of levels of both cytokines indicated that non-targeting SLS NPs failed to exert stable long-term anti-inflammatory effect. Vancomycin has been long taken as the first line antibiotics against MRSA related infection.19 However, as we have tested, the anti-inflammatory outcome was sub-optimal since both of the cytokines we tested gradually increased post vancomycin injection, in line with previously reported.20 Several factors might be responsible for this, we mainly envisioned that the limited blood circulation time due to their low molecular weight hindered it from accumulating in lung lesions in enough amount.21 Increasing the injection dosage might enhance the therapeutic potential of vancomycin at the risk of inducing vancomycin resistant pathogen. However, by taking advantage of our platform, such drawbacks could be safely avoided. In Fig. 6B, typical images of lungs (H&E staining) post treatment were given. Abundant diffusion of inflammatory cells like neutrophils or pro-inflammatory macrophages are representative indicators of inflammation. It could be clearly seen that the amount of neutrophilic granulocytes infiltrated in lung stroma or alveolar (blue stained) was significantly reduced post targeting SLS NPs treatment. Loss of tracheal epithelium cilia structure (highlighted from the images with low resolution) and prevalence of inflammatory cells post non-specific SLS NPs and vancomycin injection indicated that these platforms were invalid in inhibiting MRSA pathogen inhibition and alleviating inflammatory response, in consistent with the results of Elisa assays.
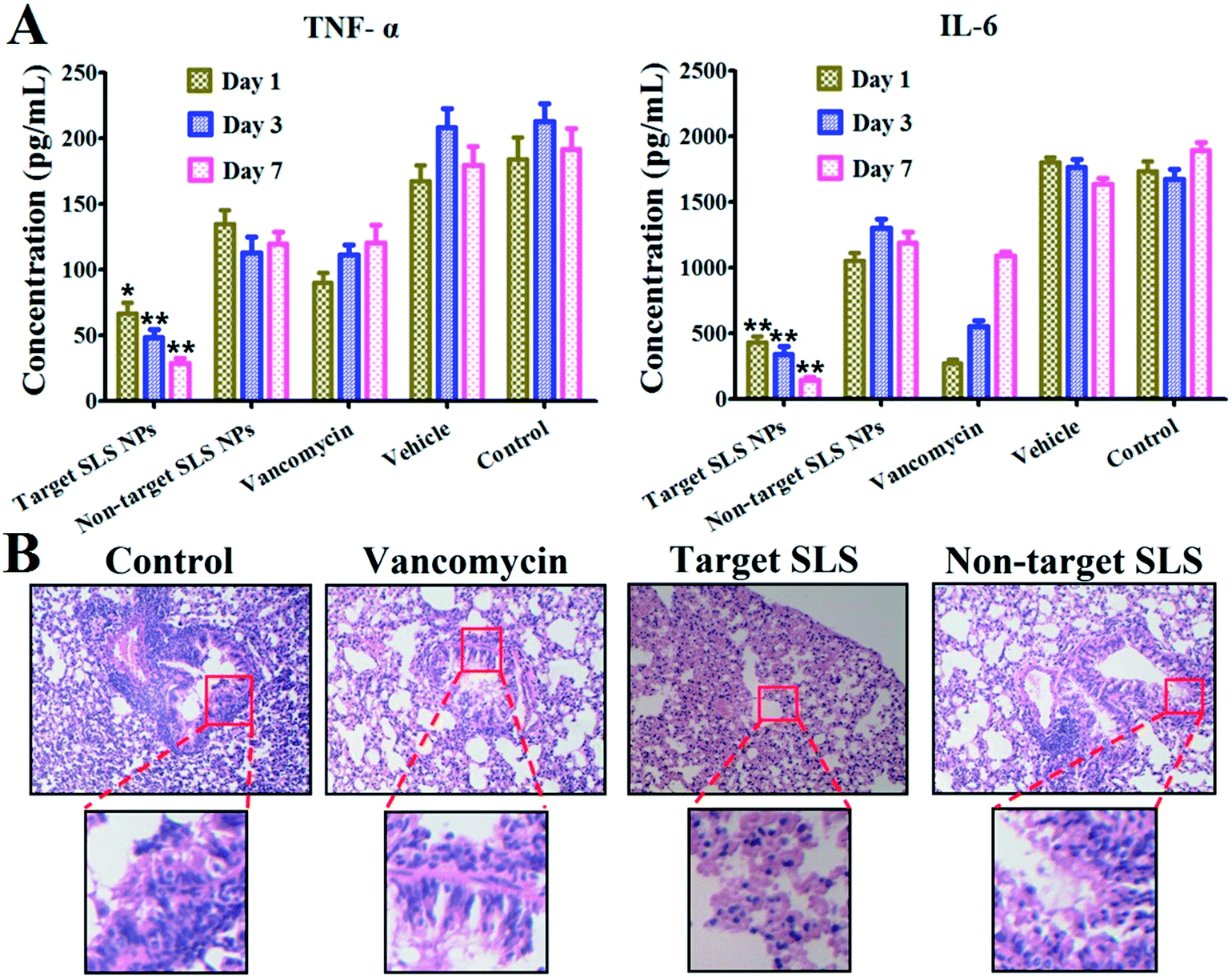 | ||
| Fig. 6 (A) TNF-α and IL-6 cytokine level in BALF of pneumonia rats verified by Elisa assay. (B) Pathological study via H&E staining (original magnification× 100) (red circle: highlighted representative sections with high magnification) (**P < 0.001; *P < 0.01). | ||
Long term safety of nanomedicine is crucial for the success of clinical translation. Thus, we verified the potential cytotoxicity of our platform in healthy rat post injection. As shown in Fig. 7, only the serum level of alanine aminotransferase (ALT, typical biomarker for liver function) and lymphocytes (LYM, cell group responsible for systematic inflammatory response) increased slightly shortly post treatment, and gradually recovered after that. Levels of blood urea nitrogen (BUN) and platelets (PLT), indicators of kidney and spleen function, were around the normal level over time, in consistent with that of that of hematological markers. Collectively, results of biochemical and hematological studies give strong evidence of the well tolerance of targeting SLS NPs for in vivo application.
 | ||
| Fig. 7 Serum biochemical and hematology assay of normal rats post targeting SLS NPs treatment over time. | ||
In conclusion, we synthesized a novel MRSA pathogen selective mesoporous silica NPs based theranostic platform. By taking advantage of the silver clusters, MRSA induced pneumonia lesions could be detected with high contrast under CT guidance and the inflammatory response could be significantly reduced. We hope our bio-compatible MRSA targeting SLS nanomedicine could play an important role in future clinical trials and applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81502608).References
- E. Oldfield and X. Feng, Trends Pharmacol. Sci., 2014, 35, 664–674 CrossRef CAS PubMed.
- B. Spellberg and D. N. Gilbert, Clin. Infect. Dis., 2014, 59(2), S71–S75 CrossRef PubMed.
- F. Nguyen, A. L. Starosta, S. Arenz, D. Sohmen, A. Donhofer and D. N. Wilson, Biol. Chem., 2014, 395, 559–575 CrossRef CAS PubMed.
- E. Rubinstein, M. H. Kollef and D. Nathwani, Clin. Infect. Dis., 2008, 46(5), S378–S385 CrossRef PubMed.
- B. C. Herold, L. C. Immergluck, M. C. Maranan, D. S. Lauderdale, R. E. Gaskin, S. Boyle-Vavra, C. D. Leitch and R. S. Daum, JAMA, 1998, 279, 593–598 CrossRef CAS PubMed.
- E. J. Gorak, S. M. Yamada and J. D. Brown, Clin. Infect. Dis., 1999, 29, 797–800 CAS.
- M. H. Kollef, P. Silver, D. M. Murphy and E. Trovillion, Chest, 1995, 108, 1655–1662 CrossRef CAS PubMed.
- C. H. Marquette, H. Georges, F. Wallet, P. Ramon, F. Saulnier, R. Neviere, D. Mathieu, A. Rime and A. B. Tonnel, Am. Rev. Respir. Dis., 1993, 148, 138–144 CrossRef CAS PubMed.
- J. Y. Fagon, J. Chastre, A. J. Hance, M. Guiguet, J. L. Trouillet, Y. Domart, J. Pierre and C. Gibert, Am. Rev. Respir. Dis., 1988, 138, 110–116 CrossRef CAS PubMed.
- D. Huo, J. Ding, Y. X. Cui, L. Y. Xia, H. Li, J. He, Z. Y. Zhou, H. W. Wang and Y. Hu, Biomaterials, 2014, 35, 7032–7041 CrossRef CAS PubMed.
- L. Pan, J. Liu, Q. He, L. Wang and J. Shi, Biomaterials, 2013, 34, 2719–2730 CrossRef CAS PubMed.
- C. H. Marquette, D. Wermert, F. Wallet, M. C. Copin and A. B. Tonnel, Chest, 1999, 115, 200–209 CrossRef CAS PubMed.
- J. Deckert, P. U. Park, S. Chicklas, Y. Yi, M. Li, K. C. Lai, M. F. Mayo, C. N. Carrigan, H. K. Erickson, J. Pinkas, R. J. Lutz, T. Chittenden and J. M. Lambert, Blood, 2013, 122, 3500–3510 CrossRef CAS PubMed.
- K. Yang, L. Feng, H. Hong, W. Cai and Z. Liu, Nat. Protoc., 2013, 8, 2392–2403 CrossRef CAS PubMed.
- X. Li, N. Anton, G. Zuber, M. Zhao, N. Messaddeq, F. Hallouard, H. Fessi and T. F. Vandamme, Biomaterials, 2013, 34, 481–491 CrossRef PubMed.
- E. P. Matthews, Journal of Radiological Science and Technology, 2015, 86, 623–638 Search PubMed.
- G. Z. Feuerstein, T. Liu and F. C. Barone, Cerebrovasc. Brain Metabol. Rev., 1994, 6, 341–360 CAS.
- Z. Xing, J. Gauldie, G. Cox, H. Baumann, M. Jordana, X. F. Lei and M. K. Achong, J. Clin. Invest., 1998, 101, 311–320 CrossRef CAS PubMed.
- K. Hiramatsu, Drug Resist. Updates, 1998, 1, 135–150 CrossRef CAS PubMed.
- G. Steinkraus, R. White and L. Friedrich, J. Antimicrob. Chemother., 2007, 60, 788–794 CrossRef CAS PubMed.
- T. A. Golper, H. M. Noonan, L. Elzinga, D. Gilbert, R. Brummett, J. L. Anderson and W. M. Bennett, Clin. Pharmacol. Ther., 1988, 43, 565–570 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |