
Tunable photocatalytic and photoelectric properties of I−-doped BiOBr photocatalyst: dramatic pH effect†
Abstract
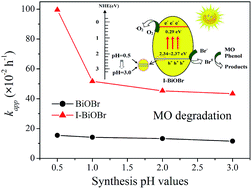
A series of I−-doped BiOBr (I-BiOBr) photocatalysts were prepared through adjusting the synthesis pH values of the reaction solution. The crystalline structure, light absorption, morphology and element component were measured by XRD, DRS, SEM, TEM, EDS, ICP-OES and XPS technologies. The doping of I− ions strongly broadened the visible light absorption range and improved the activity of I-BiOBr. More importantly, the photocatalytic activity of I-BiOBr dramatically depended on the synthesis pH values, in which I-BiOBr (0.5) displayed the best performance for removing methyl orange and phenol under visible light. The photoelectric property measurement revealed that I-BiOBr had stronger electron–hole separation efficiency than the corresponding BiOBr reference samples. With increase in the pH values, the I− ion doping contents varied a little but the surface charge state changed regularly. The decrease in the surface adsorbed negative Br− ions reduced the formation efficiency of active Br0 and resulted in low activity of I-BiOBr. This study provides a facile and efficient way to control the photocatalytic and photoelectric behaviors of novel I−-doped BiOX photocatalysts via changing the pH values of the reaction solution during synthesis.
 Please wait while we load your content...
Please wait while we load your content...

