
In search for effective and definitive treatment of herpes simplex virus type 1 (HSV-1) infections
Krzysztof Szczubiałka
a,
Krzysztof Pyrć
b and
Maria Nowakowska
*a
aFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060 Kraków, Poland. E-mail: nowakows@chemia.uj.edu.pl
bFaculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387 Kraków, Poland
First published on 17th December 2015
Abstract
Herpes Simplex Virus type 1 (HSV-1) is a nuclear replicating enveloped virus. It is considered to be one of the most frequent pathogens acquired by humans. Worldwide about 50–90% of individuals are seropositive for this virus. The symptoms of primary HSV-1 infections include appearance of watery blisters (cold sores) on the skin, tongue, lips, genitals or buccal mucosa and keratitis. HSV-1 infection is transmitted by direct contact with lesions or body fluids (typically saliva) and is incurable. Except for productive infections, HSV-1 can cause a latent infection of the host who becomes a lifelong viral reservoir vulnerable to reoccurrence of the disease. The presence of virus material may affect general health as the virus interferes in the host's cell life cycle. The viral genome stays in a latent state in the nucleus of sensory neurons of the trigeminal ganglia (TG) avoiding the host's immune system. These neurotropic and neuroinvasive properties of HSV-1 can cause life-threatening diseases like neurological disorders, encephalitis, and blindness. HSV infection is the most common cause of corneal blindness in the Western world. It is also linked to Alzheimer's disease. HSV-1 infections are sometimes dangerous for immunocompetent individuals and are very dangerous for babies and immunodeficient people. Currently, there is no drug available which would allow eradication of the virus from the infected person's body. Approved therapies for the HSV-1 infection are based on the application of inhibitors of the viral DNA replication. Although they are effective against primary infection, they cannot protect a person against the virus in the latent state resulting in the reoccurrence of the disease. The occurrence of the viral strains resistant to these drugs creates serious problems. Thus, there is a need to devise a novel approach for the development of drugs which could be effective against HSV-1, preferentially acting according to a different mechanism from those of the currently used compounds. This review presents the structure of HSV-1, the mechanisms of productive and latent infections, gives an overview of current approved therapies and proposes a novel approach to develop antiherpes agents which should be useful in these studies.
 Krzysztof Szczubiałka | Professor Krzysztof Szczubiałka received his Ph.D. from Faculty of Chemistry, Jagiellonian University, Krakow, Poland in 1996. He held a postdoctoral position at Vrije Universiteit Brussel, Brussels, Belgium, and at Osaka University, Osaka, Japan. Currently, he is a member of Nanotechnology of Polymers and Biomaterials Group at the Jagiellonian University. His scientific research is focused on polymers for biomedical and environmental applications with particular interest in polymers for the control of blood coagulation, antiviral polymers, polymeric cell culture supports, selective adsorbents for biomolecules and hybrid polymeric-inorganic photocatalysts for the degradation of water micropollutants such as antibiotics and cyanobacterial toxins. |
 Krzysztof Pyrć | Dr Krzysztof Pyrc received his Ph.D. in the field of infectious diseases at the University of Amsterdam, the Netherlands in 2007. Subsequently, he moved to the Jagiellonian University in Poland. In 2013 obtained a D.Sc. degree at the University of Lodz, Poland. Currently working mostly on molecular mechanisms of virus infection and development of novel antiviral compounds. Head of the Laboratory of Virology and Infectious Animal Facility, both working at the BSL3+ standard. |
 Maria Nowakowska | Professor Maria Nowakowska received her Ph.D. and D.Sc. degrees in physical chemistry at the Faculty of Chemistry, Jagiellonian University (JU). She was involved in long-term scientific collaboration with Department of Chemistry, University of Toronto, Canada, where she held post-doctoral and visiting professor positions in the research group of the late Professor James E. Guillet. Currently she is a chair in Department of Physical Chemistry and Electrochemistry and a head of Nanotechnology of Polymers and Biomaterials Group at the JU. Her scientific interest concentrates on materials science. |
1. Introduction
Viruses are considered to be one of the major causes of human morbidity and mortality. Viral infections have a considerable impact on the molecular physiology of the host cells, typically leading to disturbances in the expression of cellular genes, an increase in the level of stress proteins, and the activation of the innate immune system. These changes are related to the perturbation of the cell signaling networks induced by the viral infection. Viruses also are able to actively use the signaling systems of the host cells to create a favorable environment for their own replication and amplification.1–3The herpesvirus family (HSV, termed also as Herpesviridae) consists of over 100 enveloped, double stranded, neurotropic DNA viruses.4 They have a short replication cycle (about 18 h). The virus replication leads to the destruction of the host cells.5 Eight of them are infecting humans, while the rest infect animals, both domestic and wild species. The most common are HSV-1 and HSV-2 which have been classified by the International Committee on the Taxonomy of Viruses (ICTV) as the members of the genus Simplexvirus in the subfamily Alphaherpesvirinae in the family Herpesviridae.
The HSV-1 is considered to be one of the most frequent pathogens acquired by humans.6–8 The statistics show that 50% to 90% of individuals worldwide are seropositive for that virus.9 Human herpesvirus infections are endemic and direct contact with the infected person, such as kissing (contact with saliva), sexual contact, or contact with the infected lesions or blood, are the main ways of their transmission. The HSV-1 infection occurs usually in childhood. The primary HSV-1 infections are usually asymptomatic but after short incubation period of about 1 week typical symptoms occur. They include appearance of watery blisters in the skin, tongue, lips, genitals, buccal mucosa and the hard and soft palate. That viral infection is considered to be incurable. While introduced to the human/animal body, the virus successfully avoids clearance by the immune system, entering neurons in its dormant state known as latency, resulting in a lifelong infection.5,10–18 That poses the danger to the infected individual as viral reactivation may occur months or even years after clearance of acute symptoms on exposure to a number of stimuli such as ultraviolet light, immunosuppression or emotional stress. In an outbreak, the virus stored in a nerve cell becomes active and it is transported via the neuron's axon to the skin, where virus replication cycle starts and infection symptoms are visible. The virus is also dangerous for people contacting the infected person who functions as a lifelong reservoir for the virus.17–21
Transmission of the herpesvirus to newborns and immunosuppressed individuals can be very dangerous. Common clinical manifestations of HSV-1 infection are typically limited to cold sores (blisters) of the mouth and keratitis in the eyes while HSV-2 is mostly responsible for genital lesions. However, both viruses are capable of causing lesions on the same body sites and both can cause life-threatening diseases like neurological disorders, encephalitis, and blindness.22,23 Approximately 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases of corneal HSV infections are reported yearly in the US alone. There are 50000 new and recurrent cases per year24–27 with visual disability reported as high as 40%.28
000 cases of corneal HSV infections are reported yearly in the US alone. There are 50000 new and recurrent cases per year24–27 with visual disability reported as high as 40%.28
Recently, the neurotropic and neuroinvasive properties of HSV-1 have been linked with neurodegenerative diseases such as Alzheimer's disease.29–33 In the USA, herpes simplex encephalitis (HSE) is considered to be the leading cause of sporadic lethal encephalitis in immune-competent individuals, with death rate of ∼70% of untreated patients and about 20% when anti-viral therapies were introduced.34 Additionally, more than 50% of survivors suffer from significant neurological disorders. Paradoxically, however, the neurotropic and neuroinvasive properties of the virus may have a positive aspect allowing application of HSV-1 in gene delivery into neurons35 or in the treatment of breast cancer meningeal metastases.36 Herpes simplex stromal keratitis (HSK), an immunopathological disease, is the most frequent serious viral eye infection in developed countries and is the leading cause of corneal blindness due to an infectious agent.21,24–27 Also, there is an increasing number of genital herpes cases associated with HSV-1.
2. The virus
Herpes simplex virus is a large dsDNA virus with a genome of ∼150000 bp, encompassing >90 open reading frames, encoding for 84 proteins. Genomic dsDNA is present mostly in linear form in virions, forming a torus together with polyamines – spermine and spermidine.37,38 After transfer to the nucleus and release from the core, DNA rapidly circularizes and serves as a template for transcripts production. The virus has a very peculiar genome organization, with long (L) and short (S) regions located within a single DNA molecule. These regions consist of unique elements UL and US, separated and flanked with repeats. Surprisingly, the L and S elements may be present in inverted form, together with the adjoining flanking elements.39,40 Graphical overview of the genomic DNA molecule is provided in Fig. 1.
 | ||
| Fig. 1 Scheme of HSV-1 genome organization. UL and US elements represent two genomic regions, long and short one, respectively. aL and aS elements at genome termini represent terminal repeats of the L or S region, respectively. an and am represent n or m repetitions of the a region at the L or S termini, respectively. b and c represent terminal b and c sequences of the L and S region. | ||
The genome is inserted into the icosahedral capsid, formed of four viral proteins VP5, VP19C, VP23 and VP26. The capsid is further protected by lipid bilayer, coated with glycoproteins (gB, gC, gD, gE, gG, gH, gK, gM) and transmembrane proteins (UL20, US9, and possibly also UL24, UL34, and UL43) (Fig. 2). Tegument is located between capsid and envelope and contains proteins (VP16, VP22, VP1-2, UL14, UL17, VP11-12, VP13-14, US9, US10, and US11), important for virus replication, especially on early stages.
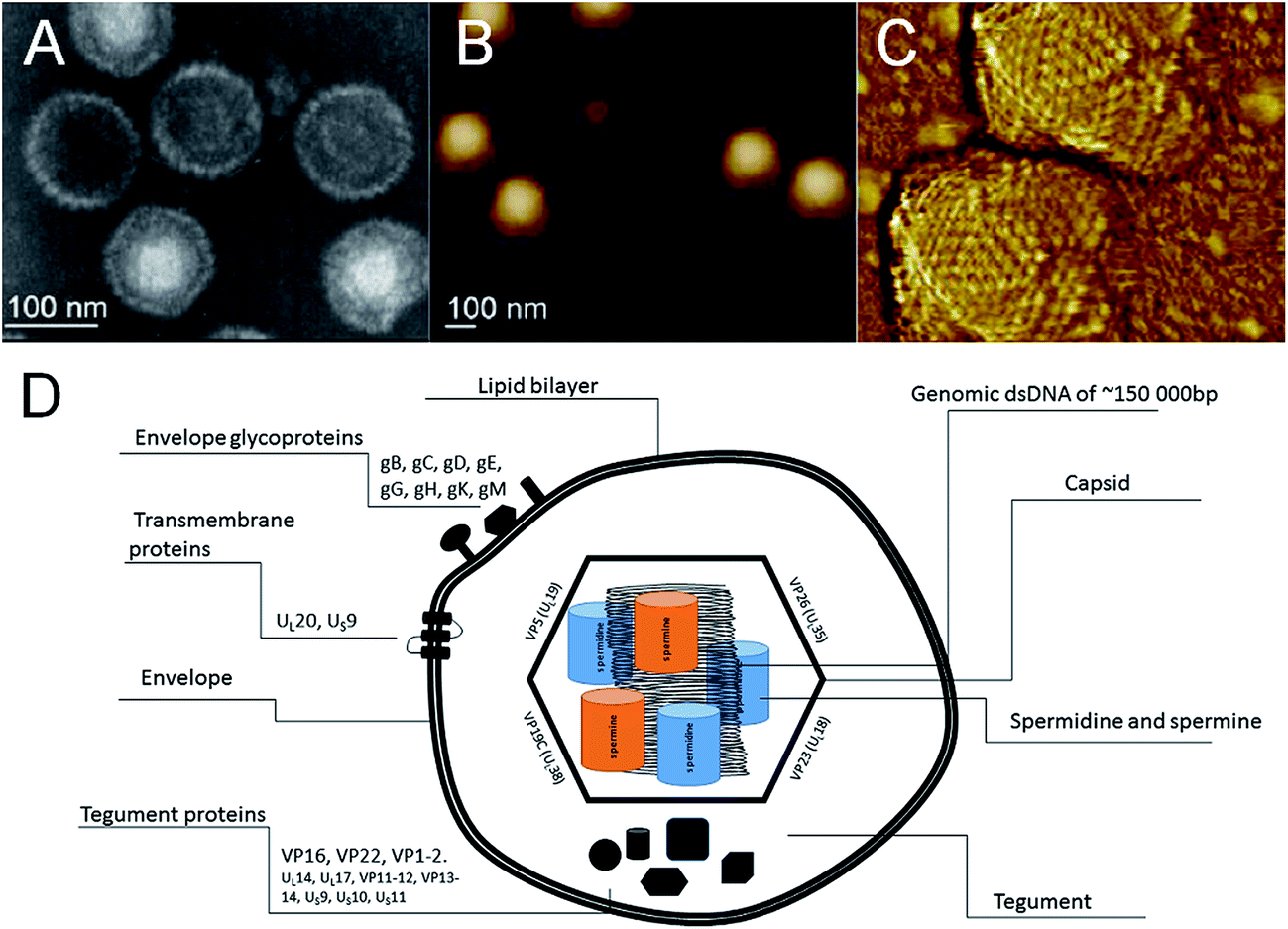 | ||
| Fig. 2 (A) SEM image of the capsid preparation. (B) HSV-1 capsids adsorbed to the surface of poly-L-lysine-coated mica (1.60 × 1.38 μm). (C) Two capsids with apparent icosahedral symmetry and clearly distinguishable triangular faces (510 × 560 nm). (D) Schematic organization of the HSV-1 virus particle. (A–C) were adapted with permission from ref. 41. | ||
3. HSV-1 replication
3.1. HSV-1 cell entry
Simplified scheme of HSV-1 replication cycle is presented in Fig. 3.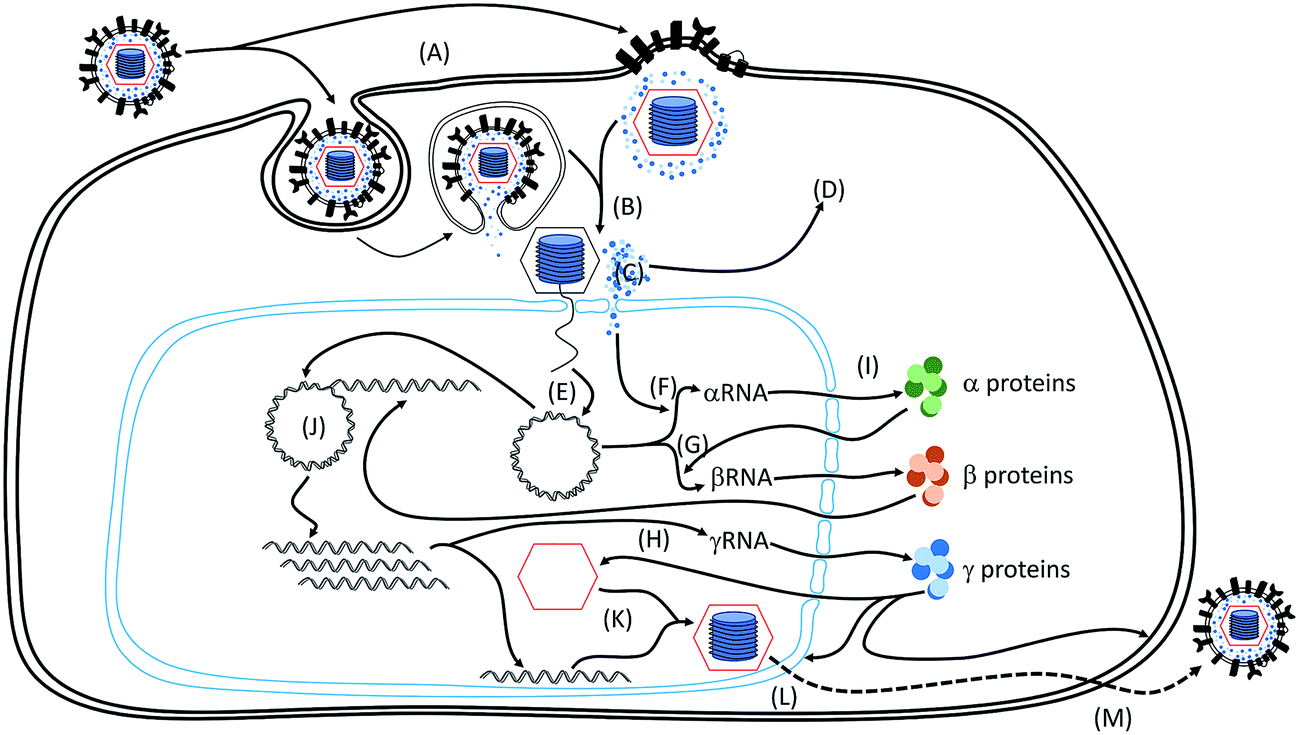 | ||
| Fig. 3 Replication cycle of HSV-1. Stages A–M are described in detail in the text. | ||
HSV-1 infection is initiated with virion attachment to the cell surface. Three virion surface glycoproteins are essential for virus–receptor interaction. First, glycoproteins B and C mediate binding to cell surface glycosaminoglycans, what enables interaction between viral glycoprotein D and entry receptors. There are three major entry receptors described – nectins, herpesvirus entry mediator (HVEM) and 3-O-sulfated heparan sulfate (Fig. 3A).42–46 The nectins belong to the immunoglobulin superfamily and are involved in cell–cell adhesion. They are present in several cell types, including epithelial cells and neurons. Nectin-1 serves as an entry receptor for HSV-1, mostly in neurons and keratinocytes. The gD binding site is located in the V-domain and overlaps a functional binding site for the natural ligands of nectin-1.47,48 HVEM is a widely expressed tumor necrosis factor (TNF) receptor superfamily member, which plays diverse roles in immune signaling.49 Although it was described to serve as the HSV-1 receptor, HVEM role seems to be limited to viral spread during ocular infections.50 The last described receptor, 3-O-sulfated heparan sulfate, is a form of heparan sulfate enzymatically modified by D-glucosaminyl-3-O-sulfotransferases. The role of this receptor is not fully understood, but it was suggested that it may play a role during cell-to-cell virus spread and viral infection of the brain and the eye.45,51,52 Binding of glycosaminoglycans by gB/gC and subsequent binding of gD to the receptor triggers a series of events, resulting in gH/gL mediated activation of gB, being the type III fusion protein.53–55 HSV-1 fusion may occur on cell surface, but the virus may also be internalized by endocytosis, and the gB mediated membrane fusion may take place in an endocytic vesicle (Fig. 3A).56,57 Fusion of the virion – cell membranes results in release of capsids and tegument proteins to the cytoplasm (Fig. 3B). Subsequently capsids are transported along microtubules to nuclear pores by dynein/dynactin motor protein complexes.58,59 Analysis of virus trafficking suggests that during translocation to the nucleus empty capsids remain outside the nucleus, while DNA is transferred into this organelle.60,61 Virions are docked to the nuclear pore by importin β and nuclear pore complex proteins, and DNA is ejected to the nucleus via the portal formed with tegument UL6 protein with assistance of several other viral proteins (Fig. 3C).62–64 Some tegument proteins also play a role in initiation of virus replication. To make an example, UL41 protein carries endoribonuclease activity and shuts down cellular machinery by degradation of host's mRNA (Fig. 3D).65 Other tegument protein – VP16 (UL48) – is translocated to the nucleus, where it induces transcription of α genes.66,67
3.2. Virus transcription
Upon translocation of HSV-1 DNA to the nucleus, it rapidly (∼30 minutes) undergoes circularization, carried out by host's DNA ligase IV/XRCC4 (Fig. 3E).68 Circular HSV-1 DNA associates with cellular and viral proteins and may serve as a template for transcription or replication.HSV-1 RNA molecules are produced by the transcription complex, formed by multiple viral proteins and host RNA polymerase II. The transcription of HSV-1 genes and subsequent expression of viral proteins is a well-regulated process. Viral genes are divided into α, β and γ groups, which are translated at different time-points of infection, increasing the process efficiency. Simplifying, while α proteins are responsible mainly for the establishment of the infection, several β proteins are involved in viral replication and structural proteins mostly belong to the γ group.
Virus transcription is initiated by de-repression of α genes promoters, and this process is mediated by VP16 protein (UL48) delivered to the newly infected cell in the tegument. VP16 complexed with octamer-binding protein (Oct-1) and host cell factor 1 (HCF-1) de-represses the α genes promoters and recruits transcriptional factors (Fig. 3F).66,67,69 The second step in transcription of HSV-1 genes encompasses initiation of β and γ1 genes transcription. This process is controlled by α protein ICP4 (α4), and, to a lesser extent, by the ICP0 (α0) protein. ICP0 accumulates in the nuclei of infected cells within 6–9 hours post infection. The protein is non-essential for replication, but it promotes viral replication in vitro and in vivo. The major function of the protein is to de-repress β and γ1 promoters by interaction with cellular factors, such as HDAC, CoREST, BMAL1 and cyclin D3.69–71 The second protein involved in progression to β/γ1 stage, ICP4, enables transcription of these genes complexing with transcription factors and recruiting RNA polymerase II. Transcription of some β genes requires also ICP27 protein (UL54), which is interacting with RNA polymerase II (Fig. 3G).69,72–76 For replication of late γ genes DNA replication is needed. Further, except for aforementioned ICP4 and ICP27 proteins, initiation of late γ genes transcription requires also ICP22 (US1) and ICP8 (UL29) proteins. ICP8 is believed to participate in recruitment of RNA polymerase II by bridging DNA to ICP27/polymerase complexes. ICP22, on the other hand, together with viral and cellular protein kinases mediates phosphorylation of several proteins involved in transcription (Fig. 3H).69,77–80 Post-transcriptionally, viral RNA is spliced and exported to the cytoplasm by viral and cellular proteins, where it serves as a template for protein production (Fig. 3I).81
3.3. Virus replication
Seven viral proteins are involved in replication of HSV-1 DNA. These include viral DNA polymerase complex, formed by the catalytic subunit encoded by the UL30 gene and the processivity factor, encoded by UL42 gene and the helicase–primase complex, formed by UL5, UL8 an UL52 proteins. Further, replication of DNA requires UL9, which binds to the origin of replication on HSV-1 genome, ICP8 protein (UL29) binding to the ssDNA and several other cellular factors. During replication, the origin binding protein binds to the oriL or oriS elements in HSV-1 genome and initiates DNA unwinding. Subsequently, ICP8 protein binds to the ssDNA, and complexes together with helicase–primase and polymerase. DNA replication initially progresses via the theta form, where replicative complexes localize at each fork. Subsequently, replication mode changes into the rolling cycle replication, which is ori independent and does not require origin binding protein. As a result, concatemeric head-to-tail genomes are formed, further cut to form mature genomes before encapsidation (Fig. 3J).82–853.4. Egress
Assembly of capsids from γ proteins occurs in the nucleus. The complete viral capsid is formed by four proteins (VP5, VP19C, VP23 and VP26). Encapsidation of viral DNA requires several viral proteins, including the UL6 protein, forming a portal of entry for the nucleic acid. A complex of three viral proteins – UL15, UL28 and UL33 – was proposed to cleave the concatemeric DNA into monomers during encapsidation (Fig. 3K). Encapsidated DNA needs to be transferred to the extracellular space, which involves crossing of two membranes and the cytoplasm barrier.86–90Herpesviruses have evolved a unique mechanism for transport through the nuclear membrane. Filled capsids bud through the inner nuclear membrane, and subsequently these enveloped capsids fuse with the outer nuclear membrane (envelopment–de-envelopment process). Envelopment seem to be promoted by viral proteins UL31 and UL34 forming the nuclear egress complex, which disrupt nuclear lamina and enable the virus budding. The envelopment itself is mediated by interaction of HSV-1 capsid/tegument proteins with membrane proteins. Subsequently, the virus is de-enveloped, which requires fusion of the capsid envelope with the outer nuclear membrane (Fig. 3L). Secondary envelopment of capsids coated with tegument proteins occurs on cellular membranes of intracellular compartments. Enveloped viruses are subsequently released from the cell via exocytosis, where vesicle containing virions are fusing with the outer membrane (Fig. 3M).91–96
3.5. Latent infection
One of the most intriguing features of the HSV-1 biology is its ability to cause latent infection, enabling the virus to remain dormant in the host. During infection, HSV-1 enters the body by mucosal surface or broken skin barrier and causes productive infection in the epithelial layer. Subsequently, the virus enters sensory neurons axons and is transported to the neurons in ganglion, where it establishes latent infection.97,98 During this phase virus replication cycle is hampered and the virus in its circular form is hidden in the nucleus. Expression of the latency associated transcript (LAT) modulates not only virus replication but also the host cell metabolism, preventing the cell from virus recognition and apoptosis.99,100 There are several mechanisms explaining the phenomenon of HSV-1 latency in sensory neurons, including expression of neuronal tissue specific repressors of HSV-1 replication (e.g., Zhangfei factor, REST), low levels of some cellular factors required for productive HSV-1 infection (e.g., Oct-1) or different subcellular localization of these factors (e.g., HCF-1) together with the repression of virus replication by viral factors.101–104Upon reactivation, viral capsids are produced and transported along the axon and viable viruses are released at the axonal termini causing productive infection in the epithelial tissue.105
4. Current therapies of HSV-1 infection and search for novel antiviral agents
4.1. DNA replication inhibitors
In the majority of the current therapies the HSV DNA replication inhibitors are applied.106 Considering the fact that viral DNA polymerase plays an essential role in new DNA synthesis and that for this enzymatic process a proper base pairing has to be ensured, the nucleoside, nucleotide and pyrophosphate analogs have been developed as anti-HSV-1 agents. Incorporation of these into a growing DNA chain stops viral DNA synthesis and leads to the termination of chain extension. A nucleoside, acyclovir (ACV, 2-amino-9-[(2-hydroxyethoxy)methyl]-6H-purin-6-one), one of the oldest antiviral drugs used for HSV-1 treatment,107 serves as a substrate for viral DNA polymerase, competing with deoxyguanosine triphosphate for incorporation into the elongated chain (Fig. 4).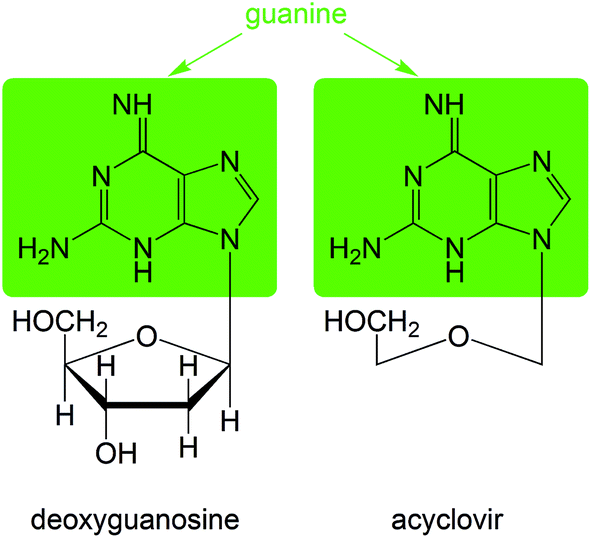 | ||
| Fig. 4 Structural similarity between acyclovir and deoxyguanosine. | ||
It was shown that ACV is activated by the viral thymidine kinase and HSV DNA polymerase to convert into ACV triphosphate for its efficient action108,109 as schematically illustrated in Fig. 5. Thus, it is predominantly active inside the viral infected cells rather than in healthy ones.
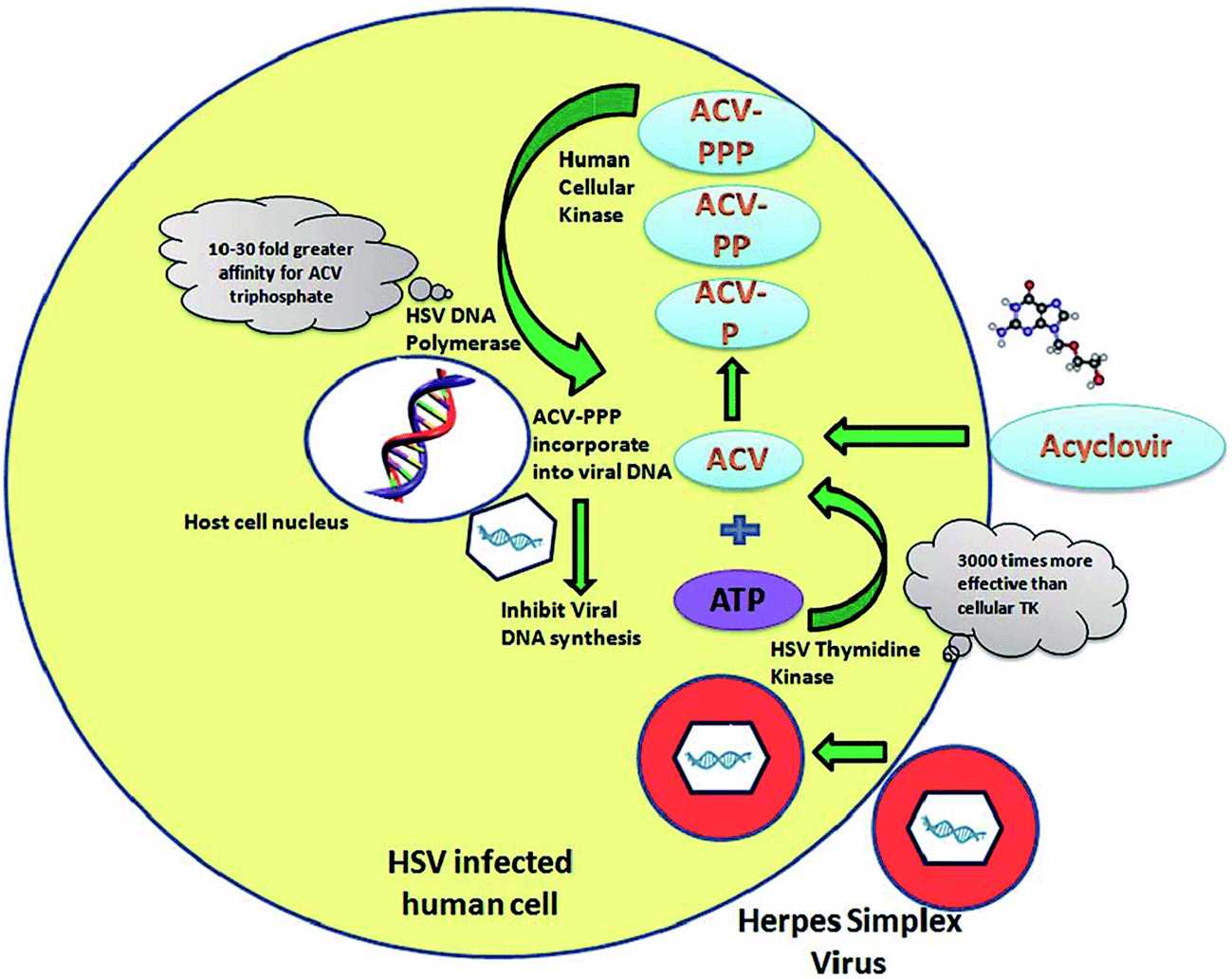 | ||
| Fig. 5 Mode of ACV action in HSV infected cells.110 | ||
ACV is an efficient DNA inhibitor with relatively low toxicity. Unfortunately, its application is limited due to its low solubility, poor permeability and limited bioavailability.107 It has been shown that the rapid biotransformation of ACV (its biological half-life is low – 2.5–3.3 h) reduces its bioavailability to less than 20%. The problem can not be solved by the increase in the dose of drug due to the saturation of absorption – it was reported that an increase in the dose from 200 to 800 mg decreased the absorption, and therefore reduced the bioavailability from 20 to 10%.111 Various approaches have been proposed to improve therapeutic action of ACV by improving its solubility, permeability and bioavailability. They mostly include development of pro-drugs and nano/microparticulate drug delivery systems allowing for prolonged and controlled delivery.
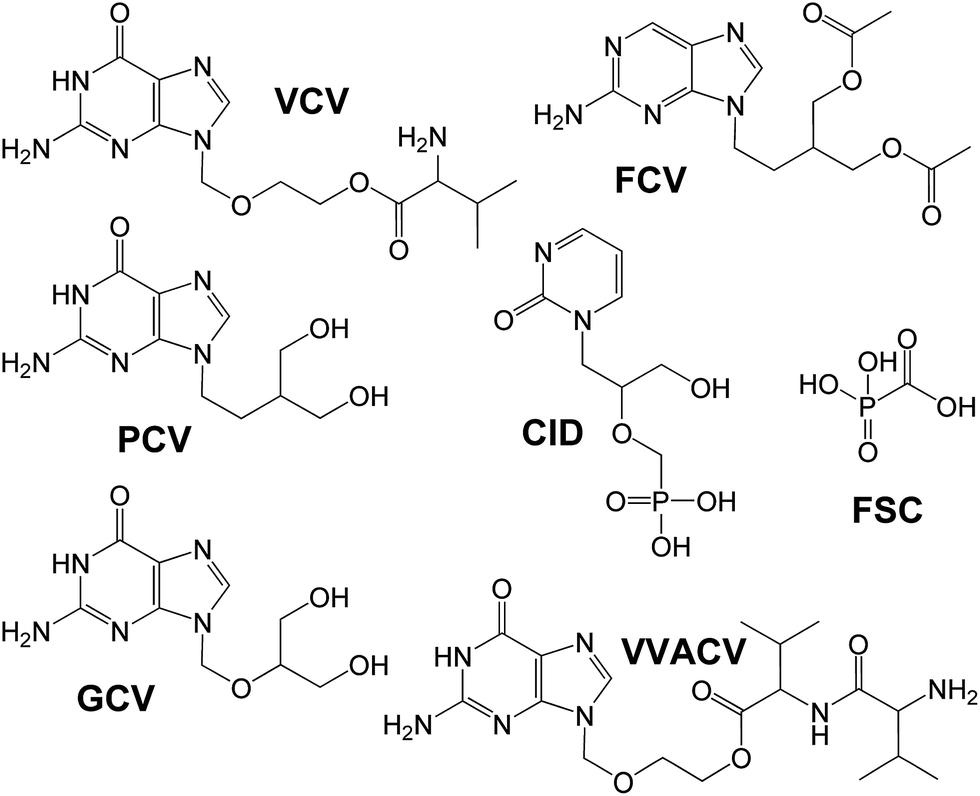 | ||
| Fig. 6 Structures of ACV prodrugs. | ||
The first nucleotide analog approved for clinical usage is cidofovir (CID), an acyclic cytidine phosphonate analog (Fig. 6). Due to the structural similarity between cidofovir diphosphate and cytosine nucleotide, it acts as a competitive inhibitor and alternative substrate for viral DNA polymerase.126 That collection of ACV pro-drugs was complemented by the highly water-soluble, characterized by low toxicity dipeptide prodrug VVACV (Fig. 6). That compound has shown excellent in vivo activity against HSV-1 in rabbit epithelial and stromal keratitis. It was suggested to be a possible drug candidate for treatment of ocular HSV infections.127 Also, a series of amino acid ester pro-drugs of ACV including L-alanine-ACV (AACV), L-serine-ACV (SACV), L-serine-succinate-ACV (SSACV) and L-cysteine-ACV (CACV) was synthesized (Fig. 7). Their affinity and permeability patterns on rabbit primary corneal epithelial cell culture (rPCEC) and on rabbit cornea were studied. It was shown that SACV could be a particularly interesting compound to be considered as antiviral ocular drug. It was stable in corneal homogenate and exhibited excellent antiviral activity against HSV-1. Due to the recognition by multiple transporters it shows higher permeability across cornea than ACV.128
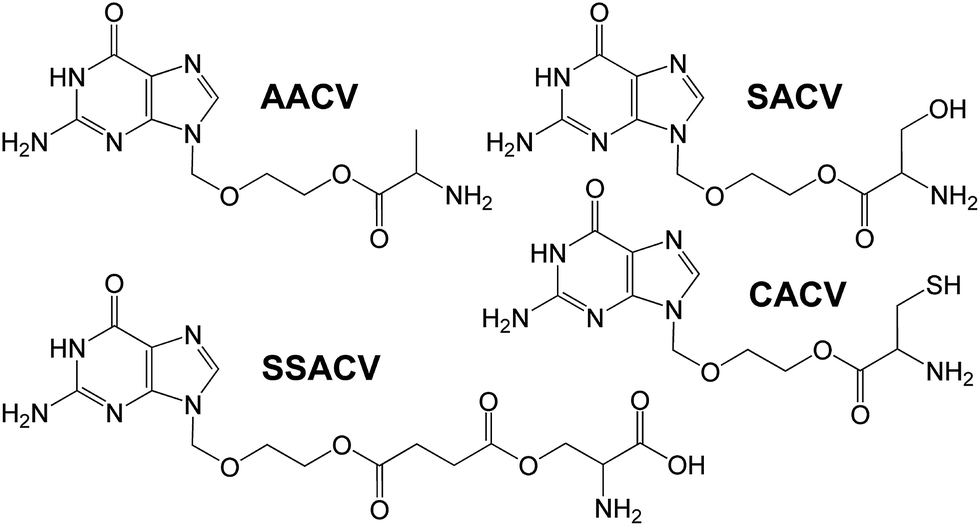 | ||
| Fig. 7 Structures of small neutral amino acid ACV prodrugs. | ||
Enhanced antiviral activity against a clinical isolate of HSV-1 was obtained using ACV loaded in nanosponge, the solid nanoparticles obtained by crosslinking of carboxylated cyclodextrin containing ACV as a drug delivery system.133 They were spherical objects with size of about 400 nm, narrow size distribution, and zeta potential equal to −38.3 mV high enough to prevent their aggregation due to the electrostatic repulsion. The ACV loading into Carb-NS reached about 70% w/w. In vitro prolonged drug release was observed with no initial burst effect. Using fluorescent nanosponge their uptake into cells was evaluated and revealed the nanoparticle internalization.
ACV-loaded chitosan-based nanoparticles were also prepared.134 The sub-spherical smooth particles with size of 132 ± 24.3 nm and dispersity of 0.159 ± 0.05 were prepared. They form stable aqueous dispersion due to electrostatic repulsion (their surface potential was 32 ± 2.87 mV). The efficiency of ACV encapsulation was 85 ± 4.38%. An in vitro release studies have shown that the process is diffusion controlled. The release profile could be fitted to Korsmeyer–Peppas model. During 48 h about 80.17 ± 2.45% of ACV was released from the nanoparticles. ACV-loaded Eudragit RLPO® nanoparticles were also prepared by the nanoprecipitation method using Pluronic F68 as a stabilizer.135 The nanoparticle average particle diameter was within the range of 82 ± 4 nm to 532 ± 5 nm, depending on the drug-to-polymer ratio and on the surfactant concentration. The highest drug entrapment efficiency reached 79 ± 2%. The sustained in vitro ACV release in pH 7.4 phosphate buffer for 24 h was observed. It was concluded that the system can be used for sustained ACV release.
The nanoemulsion hydrogel based on clove oil and castor oil (3:1 ratio), Tween 80 and Span 80 (3:1 ratio), propylene glycol and PEG-6 oleate (Myo-6V) at 3:1 ratio, can serve as an example.136 A pharmacokinetic study of the optimized ACV nanoemulsion hydrogel was conducted in vivo in rats. The relative bioavailability of the optimized ACV nanoemulsion hydrogel was improved to 535.2% and 244.6% with respect to the raw ACV hydrogel and marketed cream, respectively. The ex vivo experiments demonstrated that the obtained system showed a twofold and 1.5-fold higher permeation percentage than the control gel and marketed cream, respectively.
The nature of the HSV-induced disease stimulated an interest in developing local drug delivery systems and/or dedicated to the defined organ. They include creams, gels and implants. Zovirax® cream is currently on the market (approved by FDA in 2002) for the treatment of recurrent herpes labialis (cold sores) in adults and adolescents.137 It is a topical dermatological product containing 5% w/w of ACV and the following inactive ingredients: cetostearyl alcohol, mineral oil, poloxamer 407, propylene glycol, sodium lauryl sulfate, water, and white petrolatum. Recently, various approaches to develop the generic product were undertaken involving the studies on the novel performance matrix.
Oral mucosal delivery system for local and systemic delivery of ACV was also prepared. Temperature and pH sensitive in situ gel containing about 2% w/v of ACV was prepared using poloxamer 407, Carbopol 934, hydroxypropylmethyl cellulose (HPMC), Tween 80–ethanol (1:2 ratio), and glycerin as drug dissolving solvents.138 The sol–gel transition temperature was within the range of 28–38 °C and could be controlled by pH and polymer concentration. The mucoadhesive strength measured as detachment stress from the porcine buccal mucosa showed that the adhesive properties of gel formulations increased with the increase in the concentration of Carbopol 934. The sustained ACV release profile obeying the Higuchi kinetic model was achieved.
To ensure the achievement of desired concentration and duration of ACV release at the site of infection to minimize the adverse effects the nanoparticle based controlled release systems were developed. Stable nanoparticles 100 nm in size with surface charge of +26.1 mV were prepared from hybrid polymer blends of polyvinyl pyrrolidone with ethyl cellulose or with Eudragit RSPO.139 The efficiency of drug entrapment was optimized to 80%. ACV loaded nanoparticles showed immediate sol–gel transition at 35–37 °C. Ex vivo experiments have shown that nanoparticles were spontaneously uptaken by the human corneal epithelial cell lines and that the permeability through cornea was considerably higher than through the stomach and rectal membranes. When using nanoparticles, 10-fold reduced oral dose of ACV was required to achieve its steady-state plasma concentration needed for once-daily therapy. Additionally, the stable foam formulation (spray) was developed for site specific drug administration/action via in situ gel formation. The system can be optimized to achieve a predicted release profile under various routes of delivery.
Considerable interest has been observed in the development of ocular anti-viral drug delivery systems. That is related to the fact that herpes simplex virus infection is the most common cause of corneal blindness in the Western world. As the currently used methods are not effective in supplying drug to the cornea to prevent reactivation of viral disease there is a search for more effective delivery systems. The system for topical administration of ACV was obtained by drug incorporation into positively charged liposomal formulation.140 The pharmacokinetic profile of the drug in the aqueous humor of rabbits after topical administration was determined. Liposomal drug dispersion gave significantly higher drug concentration profile with respect to the reference aqueous dispersion formulations containing the same ACV concentration, and showed a 90 minute plateau. It was possible to obtain the drug concentration being in the upper range of the ID50 for HSV-1.
Nanosuspensions (NS), the colloidal dispersions for ophthalmic delivery of ACV, were also proposed. The system containing 3% Pluronic® F68 and 0.5% Tween® 80 allowed dispersion of particles with a mean diameter of 21.62 ± 1.07 nm with dispersity of 0.304 and a zeta potential of −19.1 mV.141 Dispersion was stable for six months. The average entrapment efficiency of ACV was 85.85 ± 3.40%. Determined in vitro release profile has shown that approximately 40.08% of ACV was released over three hours. Optimum ACV NS formulation; F7; proved itself safe when tested histopathologically on the rabbits' eyes containing 3% Pluronic® F68 and 0.5% Tween® 80. F7 showed a mean particle diameter of 21.62 ± 1.07 nm with dispersity of 0.304 and a zeta potential of −19.1 mV. It also showed an average entrapment efficiency of 85.85% ± 3.40% with approximately 40.08% of ACV being released in vitro over three hours. The in vivo studies carried out on the rabbits' eyes have shown that nanosuspension ACV formulation is safe for ophthalmic delivery. However, in vivo application of all above mentioned ophthalmic delivery systems is problematic because carriers are rapidly cleared from the ocular surface by tearing and blinking. To solve that problem the incorporation of drug into contact lenses prepared from poly-2-hydroxyethyl methacrylate (p-HEMA) hydrogels was suggested.142
In the case of HSV-related vision loss the treatment involves transplantation of donor corneas. In order to prevent the viral reactivation during surgery the implant, based on ACV containing silica (SiO2) nanoparticles incorporated into biosynthetic alternatives to donor corneas, was introduced.143 The incorporation of NPs did not affect optical clarity of the collagen-based corneal substitutes nor their biocompatibility. The sustained release of ACV from these biosynthetic implants for over 10 days was observed. The level of drug released was sufficient for effective prevention of virally-induced cell death which was not achieved when the drug was just incorporated within the hydrogel. In the latter case the rapid and complete release occurred within 24 hours. It was concluded that such biomimetic corneal substitutes with incorporated silica nanoparticles encapsulating ACV can serve as future alternatives to human donor tissue grafts, for transplantation of HSV-infected corneas.
4.2. Low-molecular-weight non-nucleoside compounds as possible HSV-1 antiviral agents
ACV and the listed above pro-drugs, which function as DNA replication inhibitors, have been shown to reduce the duration of infection and alleviate some of its symptoms, however, they are not able to eliminate the viral latency. They are processed to nucleoside analogues that are selectively activated in virus-infected cells and then block viral DNA replication. There is also a serious problem related to the fact that the treatment of HSV infections with the above-mentioned antiherpes drugs has resulted in the emergence of resistant viral strains. Resistance to them can be developed by mutations in the viral thymidine kinase and/or DNA polymerase,144 with incidence rates of 0.3–7.1%.145,146 Additionally, nephrotoxicity can occur following prolonged ACV treatment.147 Recently, a series of new non-nucleoside analogs (NNA), α-pyrone carboxamide propanol derivatives, capable of selective inhibition of viral DNA polymerase, have been synthesized and studied as HSV-1 antiviral agents.148 The systematic experimental and computer modeling studies (structure based optimization) allowed identification of a compound belonging to that series, namely, N-(3-hydroxypropyl)-2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carboxamide (PPC) (Fig. 8) as a possible anti-HSV agent. Modeling studies indicated that this compound can use the tubular cavity-A for binding to HSV DNA polymerase. Biological studies, including MTT tests and plaque reduction assay, demonstrated that this compound inhibited HSV-1 infection in a dose-dependent manner and >99% inhibition was achieved at 18 μg mL−1.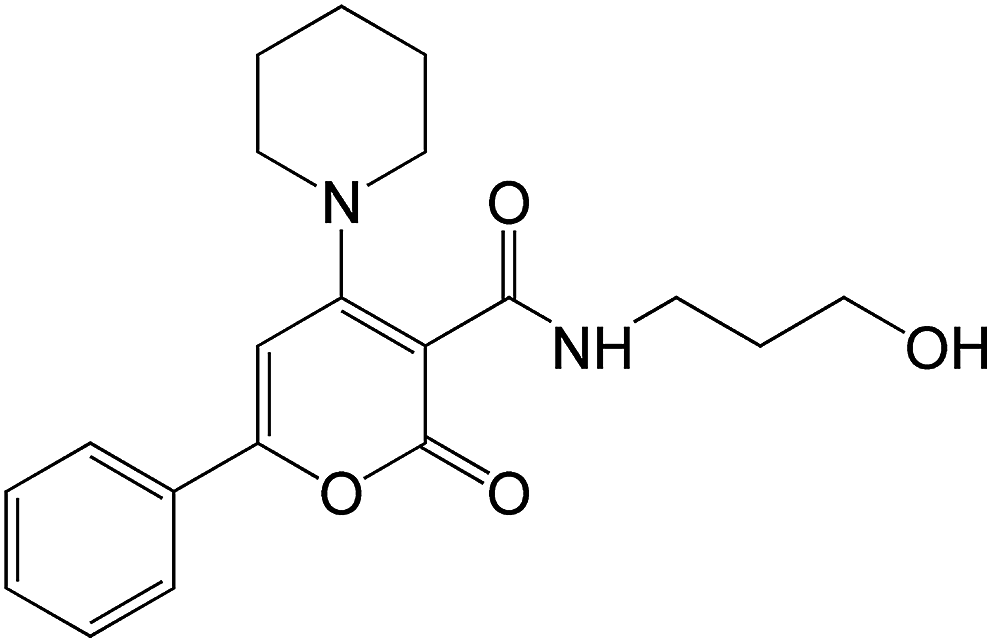 | ||
| Fig. 8 Structure of PPC. | ||
The kinetic studies have shown that the addition of studied compound to virus infected cells at 4–6 h post-infection is highly effective in killing the virus. However, it was observed that compound was unable to prevent the attachment and penetration of HSV. It was thus concluded that the mechanism of it action does not involve the prevention of viral adsorption or penetration, but rather the interference with early events of HSV replication. Based on the observation of the strong synergistic effects of the compound studied and ACV it was suggested that it may act through similar targets but at different time points.
It was also demonstrated that PPC interferes with the IE gene expression events – the reduced expression of both ICP4 and ICP27 in HSV-infected and treated with PPC cells was observed which was not seen in untreated ones. Additionally, the immunoblotting studies have shown that there was a depletion of viral DNA polymerase expression in HSV-1F infected cells treated with PPC which suggested that it can prevent HSV multiplication.
N-substituted pyrrole-based compounds having in their structures an aromatic ring with at least one hydrogen-bonding donor and acceptor group have been synthesized and evaluated for activity against different types of DNA and RNA viruses.149 Compound 1 has shown modest anti-HSV-1 activity in HEL cell cultures (EC50: 27–40 μM) (Fig. 9).
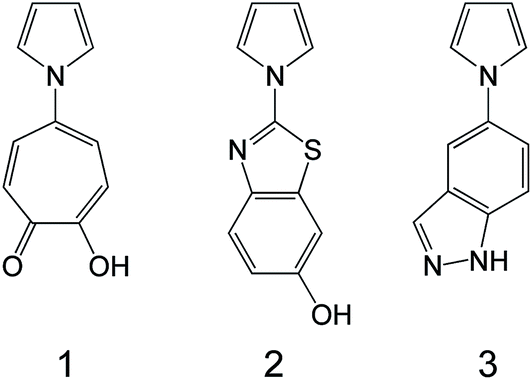 | ||
| Fig. 9 N-substituted pyrroles used for the preparation of antiviral scaffolds.149 | ||
New family of steroidal compounds derived from the endogenous hormone pregnenolone have been synthesized and tested for their in vitro antiherpetic activity.150 Studies showed that some of them inhibited remarkably spread of both HSV-1 wild type and HSV-1 acyclovir-resistant strains. These compounds interfere with the late steps of the viral replication cycle reducing infectious virion formation. It was, however, pointed out that to answer the question whether these compounds could be considered as candidates for antiherpes drugs, the controversy on the role of mammalian steroid hormones in triggering viral reactivation should be resolved.151
Recently novel anti-HSV-1 compounds based on non-nucleoside agents found earlier to have lysosomotropic activities have been tested using conventional and human induced pluripotent stem cell-derived neuronal (iPSC-neurons) cultures. Their antiviral potency and toxicity were compared with those of ACV. In vitro tests have shown that 30N12, 16F19, and 4F17 (Fig. 10) at the micromolar concentrations display the antiviral activity comparable to that of ACV (50 μM) during HSV-1 infections, reduced the number of viral DNA copies, and reduced the level of some selected HSV-1 proteins. Their antiviral activity was linked with their influence on one or more stages of the early phase of the HSV-1 lytic cycle and late stages of infection. They were shown to efficiently block events at or before DNA replication. These compounds also inhibited the reactivation of HSV-1 infection established in iPSC-neurons. The experiment was carried out in such a way that the studied compounds had not affected HSV-1 entry in tested Vero cells and neurons (the compounds were added to cell cultures 2 h after HSV-1 infection, when most of the nucleocapsids were expected to be already transported to the nuclear membrane). Considering that HSV-1 viral entry in neurons was reported to be pH-independent the lysosomotropic effect was excluded.152
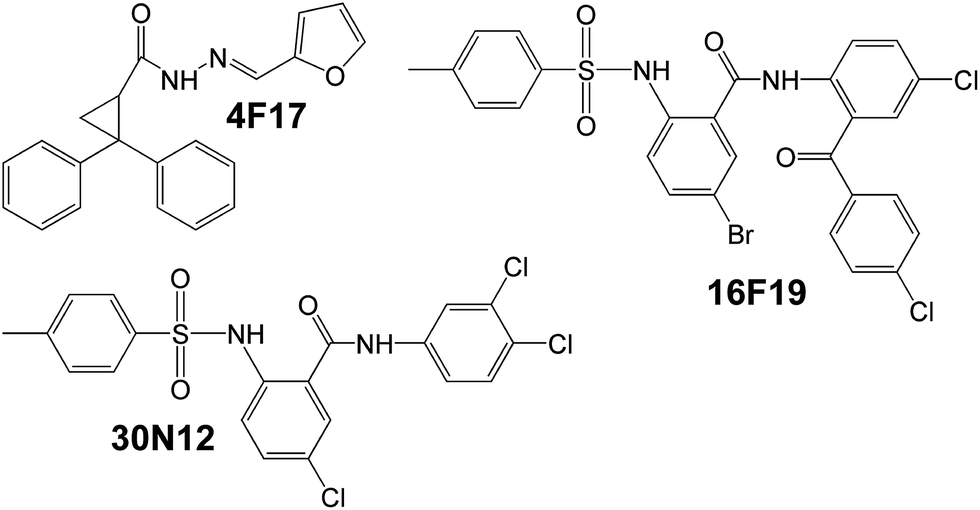 | ||
| Fig. 10 Structures of non-nucleoside antiherpes agents.153 | ||
The lysosomotropic drugs like chloroquine (CQ) and bafilomycin A1 (BFLA) have been shown to inhibit HSV-1 infections.154 The mechanism of their action is still not fully resolved although they have been shown to increase intracellular pH and most likely inhibit viral packaging and maturation through the trans-Golgi network.155
Despite the promising results of the studies on compounds that interfere with the interactions of the virus with the membrane of host cell, there is currently only one drug of that kind on market, i.e., n-docosanol,156,157 which is a saturated 22-carbon aliphatic alcohol. This alcohol dissolves in the cell membrane resulting in its modification. The changes induced in the regions used by the virus to enter the cell considerably inhibit the fusion.158 It was shown that n-docosanol inhibits fusion of the HSV-1 envelope with the cell membrane. The process involves a time-dependent metabolic conversion of the compound and results in reduced expression of viral genes in n-docosanol-treated cells. About 70% reduction in expression of a reporter gene regulated by a constitutive promoter inserted into the viral genome was observed. It was also found that virion-associated regulatory proteins release, which is an immediate post entry event, was also inhibited. Thus, inhibition of fusion between the cell membrane and the HSV envelope, and the subsequent lack of replication events, are suggested as the predominant mechanism for the anti-HSV activity of n-docosanol.
4.3. Macromolecular antiherpes agents
Macromolecular compounds, polymers in particular, are an interesting class of potential antiherpes agents because of their ability to establish strong multivalent interactions, which can be collectively much stronger than monovalent interactions.159 Of special importance from the point of view of antiviral properties are the interactions of the polymers with the virions and with the cellular receptors used by the viruses to enter the cells. The former may be established between HSV virus and polyanions mimicking heparan sulfate proteoglycans (HSPGs) present in the receptors used by these viruses. However, in spite of the encouraging preclinical results many polyanions failed in clinical trials.160 On the other hand, polycationic macromolecules, which have been less investigated as antiviral compounds, may electrostatically interact both with the negatively charged cell membrane, preventing virus adsorption onto cell surface as well as with the viral envelope, thus directly inactivating the virus particle.Polycations. Poly(amidoamine)s substituted with agmatine groups were found to show a broad antiviral activity against several viruses including HSV. This activity was ascribed to the polycationic nature of the polymer imparted by the presence of the guanidine groups.161 Another polycationic polymer Eudragit® E100 (E100), a copolymer consisting of 1
:2:1 ratio of methyl methacrylate, N,N-dimethylaminoethyl methacrylate, and butyl methacrylate, showed a membrane-destabilizing effect probably due to the presence of hydrophobic butyl and methyl residues in its structure.162 This membrane-destabilizing activity of E100 was responsible for the inactivation of a series of enveloped viruses, including HSV, occurring even in the presence of proteins.162,163 A reduction in HSV infection of over 105.0 TCID50 mL−1 was observed after 30 min of exposure to E100 at 10 mg mL−1 with the highest inhibitory effect after 60 min of incubation at 25 °C. Addition of a protein (human serum albumin, HSA) induced a dose-dependent delay in the reduction of the infections, suggesting only a transient protective effect of the protein on this virus. The incubation time had to be increased in order to attain a similar inhibitory effect at 8 °C as compared to that observed at 37 °C in the presence of the polymer. These results suggested that the major barrier for the reduction of virus infectivity is substantially kinetic rather than thermodynamic. Moreover, it was concluded that both electrostatic and hydrophobic interactions contribute to the polymer–virion interactions that determine an impairment of virus infectivity and that electrostatic interactions between E100 and HSV are prerequisites for the establishment of the hydrophobic interactions involved in the destabilizing activity on virus membranes. The interaction between E100 and the virus components was strong enough not only to limit virus infectivity but also to allow removal of the virions by E100 precipitation or by cation-exchange chromatography. The fact that the polymer could be removed by passage through cation-exchange resin indicated that E100 remained positively charged after its interaction with viral particles.
Polyanions. Poly(sodium styrene-4-sulfonate) (PSSS), a well-known synthetic polymer, was found to show anti-HSV activity.164 PSSS of the molecular weight of 500
000 Da was found to induce 3-log reduction in HSV titer at the concentration of 3 μg mL−1.165 The polymer exerted its antimicrobial activity with no evidence of cytotoxicity to cells used in the assays for HSV. It produced similar inhibition of clinical isolates of HSV-2 and was active against HIV-1.A structurally similar polymer, poly(4-styrenesulfonic acid-alt-maleic acid) (PSM) with two types of anionic groups: carboxyl groups (pKa 1.9 and 6.9) of maleic acid and styrenesulfonic groups (pKa 0.7), exhibited in vitro inhibitory activity against HIV-1, HSV-1 and HSV-2.166 PSM was likely an entry inhibitor that disrupted viral attachment to the target cells.
There are some approaches to develop antiviral therapy by specific targeting of glycoprotein–receptor interactions to inhibit the initial steps of viral entry, attachment, and fusion. The studies have shown that using peptides or oligonucleotides to interfere with virus–cell interactions via gB or gD-receptors one can inhibit the viral infection.156,169–172 There is, however, a concern regarding the low bioavailability of these compounds, their limited stability, and the high cost of the procedure. Identification of HSPGs on cell surface as the entry HSV-1 receptor was crucial in search for more specific antiviral agents having the ability to block virus attachment to that cell region on envelope glycoproteins.173–178
The other approach for HSV antiviral drug development is based on HSV fusion inhibitors. It is oriented towards blocking fusion of the virus with the cell membrane. The studies on compounds which can function as the non-specific glycoprotein–receptor targets resulted in the identification of the protein, cyanovir-N (CN-V), originally isolated from the cyanobacterium Nostoc ellipsosporum, which has been shown to act as an efficient inhibitor of HSV-1 entry into natural target cells. The inhibition of cell entry was independent on usage of HSV-1 gD receptor. It was suggested that CV-N can not only block virus entry to cells but also significantly inhibits membrane fusion mediated by HSV glycoproteins.179–183
Some hyperbranched synthetic well-defined molecules – peptide-derivatized dendrimers, with the peptidyl branching core and covalently attached surface functional peptide were suggested as promising antiviral agents directly inhibiting HSV-1. Studies on the mode of their action revealed that they prevented HSV-1 attachment to target cells due to the binding to the glycosaminoglycan moiety of the cell surface HSPGs, thus blocking virion attachment to the target cells. It was suggested that these dendrimeric polycations, containing positively charged amino acids, interact with the negatively charged carboxyl/sulfate groups of the HS receptor. These compounds exhibited highly synergistic activity with ACV.184
Recent in vitro studies have demonstrated that a p5 + 14 peptide exhibited ∼80% reduction in murine cell infection with HSV-1.185 That effect was explained as resulting from the binding of Lys residues containing peptide to the negatively charged, cell-surface HSPGs, which serve as the attachment receptor.
Among the sulfated polysaccharides, heparin has long been known to inhibit the infection of cultured cells by HSV188 but not by many other viruses (e.g., coxsackie, measles, mumps, some strains of parainfluenza and polio).189 After complexation of heparin with protamine sulfate the cytopathic effect occurred earlier indicating the neutralization of inhibitory activity of heparin together with its anticoagulatory action.188 The results on the use of heparin for the prevention and treatment of herpetic keratitis in the rabbit, however, were disappointing.
It was found that soluble heparan sulfate and chondroitin sulfate neither enhanced nor inhibited HSV infection.190 In another study heparan sulfate was highly effective in vitro, but was not protective in vivo.164 This is an interesting finding taking into account heparan sulfate is a natural cell surface receptor for the virus.
Dextran sulfate (DS), which normally inhibits infection of cells by enveloped viruses including herpesviruses, stimulated HSV-1 infection in mouse L-cell mutant cell line defective in the biosynthesis of glycosaminoglycans, and therefore resistant to HSV infection. This occurred probably by binding stably to the cells, where it acted as a matrix for subsequent virus adsorption and entry.190
4.4. Natural antiherpes agents of plant origin
There is a growing interest in antiviral agents based on the compounds which can not only hinder the binding site on the cell but also can reduce HSV-1 protein expression by interfering into the replication process or by inducing production of antiviral cytokines in the infected cell. Interestingly, such mode of action is displayed by the compounds of natural origin. The extensive literature review on the anti-HSV herbs, extracts, fractions and compounds from natural sources was published in RSC Advances in 2013.198 Thus, in a current review we will only refer to the main findings, especially these presented in the very recent publications.Hydrolyzable tannins have been shown to inhibit binding of various HSV-1 glycoproteins to the cell surface glycosaminoglycans. They are believed to be promising candidates for antiviral agents which can stop viral cell-to-cell spread.199 Various bioactive substances obtained from plants such as anthraquinones,200 flavonoids,201 catechins,202 phloroglucinol,203 polysaccharides,204 and triterpenes114,205 have been also shown to have some viral inhibitory activities. Among natural antiviral agents the sulfated polysaccharides have been studied quite frequently. The studies included carrageenans, xylofuranan sulfate, ribofuranan sulfate, and dextran sulfate.206–214 Their modes of action have been attributed to the blockage of some early stages of the virus replication cycle and stimulation of the host immune system.202,204
Recently, the HSV antiviral activity of soybean isoflavonoids, especially daidzein, genistein, glycitein, and coumestrol have been studied (Fig. 11).215 Genistein and coumestrol inhibited HSV-1 replication and, what is particularly important, they affected not only the ACV-sensitive (KOS) strains but also the ACV-resistant (29R) strains. The mechanisms of their action were studied. It was found that these compounds did not show virucidal activity. Coumestrol interferes with various early stages of viral infection by interacting with the binding site on the cell and both compounds were able to reduce HSV-1 protein expression. Daidzein and glycitein have not shown antiviral effects against HSV viruses. It was also suggested that polyphenols present in the extracts of mung bean sprouts have potent, comparable to those of ACV, antiviral and to a lesser extent, prophylactic activities against HSV-1.216 When discussing the mechanism of these activities the MBS potential to remarkably induce antiviral cytokines in human cells was indicated.
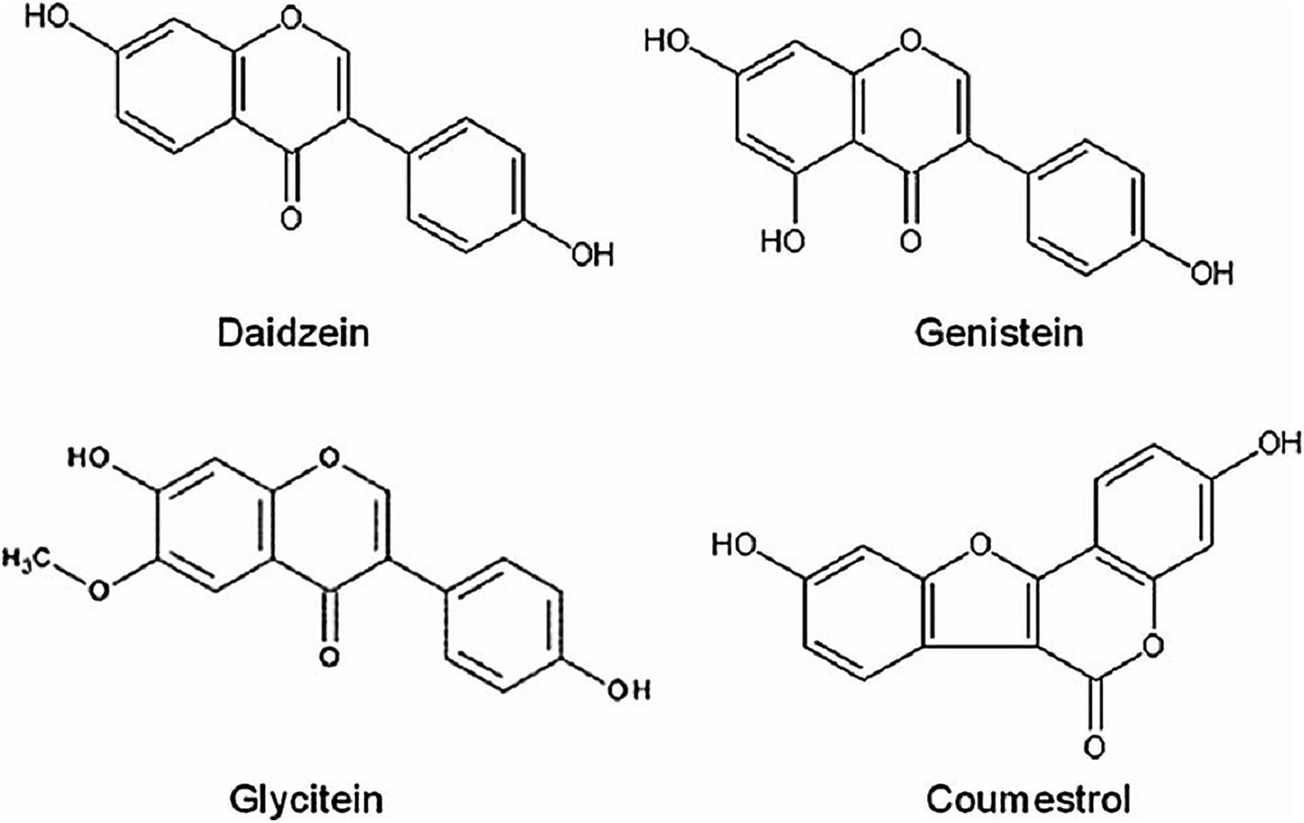 | ||
| Fig. 11 Structures of soybean isoflavonoids. | ||
The Tanacetum vulgare rhizome extracts have been shown to demonstrate the inhibitory effect on HSV-1 in vitro replication.217 Spiroketal-enol ether derivative (E)-2-(2,4-hexadiynyliden)-1,6-dioxaspiro[4.5]dec-3-ene (SE, Fig. 12A) was identified as the most active compound of the extract responsible for the inhibitory effect. This effect involved some inhibition of virus penetration but, most importantly, the compound specifically targeted the expression of viral genes, a cell-independent key regulatory step of virus replication cycle, leading to the decrease of viral protein accumulation within infected cells.
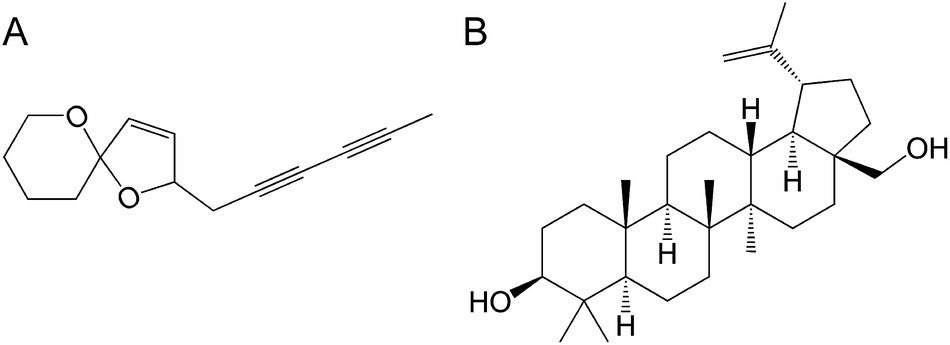 | ||
| Fig. 12 Structure of (E)-2-(2,4-hexadiynyliden)-1,6-dioxaspiro[4.5]dec-3-ene (A) and betulin (B). | ||
Betulin, a pentacyclic triterpenoid (lup-20(29)-ene-3β,28-diol), isolated from the bark of Betula genus not only displayed anti-HSV activity but also synergistic effect with ACV (Fig. 12B).218 The effect was strong or moderate depending on betulin concentration. At the concentrations lower than 0.4 μg mL−1 the additive effect was observed. What is important, triterpenoids show activity against both ACV-sensitive and ACV-resistant HSV strains. To increase the bioavailability of triterpenoids the water-soluble ionic derivatives of betulinic acid were recently obtained and are shown to retain high antiviral activity.219
Natural product, alkaloid harmaline (Fig. 13A) extracted from O. nicobarica Indian plant has been shown having potent anti-HSV-1 activity. The in vivo studies have shown that harmaline significantly reduces virus replication in mice. The compound interferes with the viral immediate transcription events.220
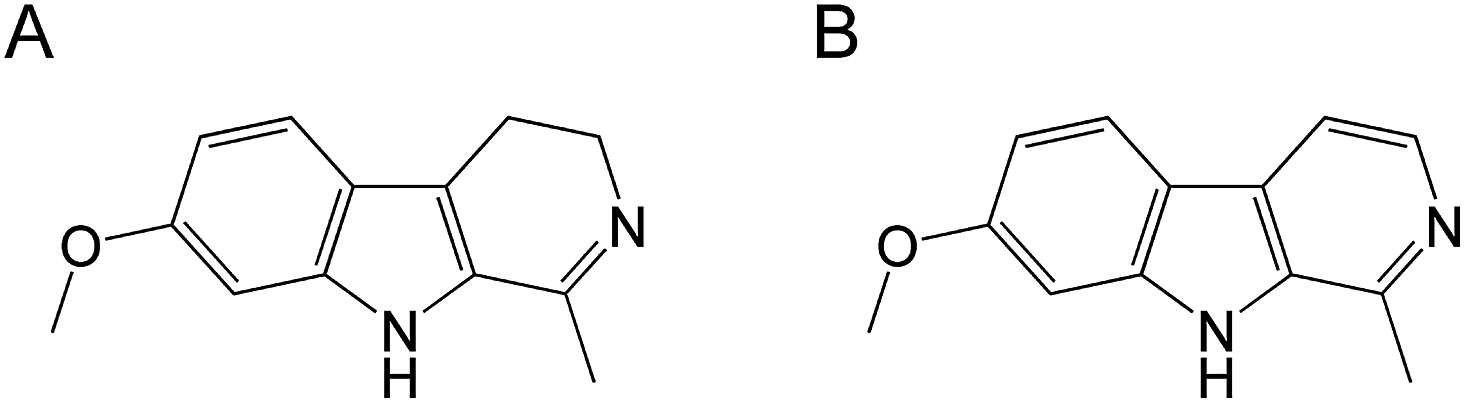
More recently, the very similar natural compound, harmine (Fig. 13B), a beta-carbon alkaloid with an indole core structure and a pyridine ring was tested as an HSV antiviral agent.221 The in vitro tests have shown that it exhibited strong antiviral activity limiting the replication of HSV-1 and HSV-2 in a dose-dependent manner at non-cytotoxic concentrations. It was observed that harmine most likely acted on post viral entry and did not directly inactivate the virions as the pre-treatment of the virus had no impact on the viral infectivity. The exact mechanism of harmine action against HSV-1 was not explained but for HSV-2 it was linked with downregulation of cellular NF-κB and MAPK pathways induced by oxidative stress following the viral infection.
One more approach to HSV antiviral drug development is based on search for microbiocides, the compounds that directly interact with the virions before they penetrate the cell. Recently, it has been shown that theaflavin polyphenols extracted from black tea derived from the leaves of Camellia sinensis plant, in particular theaflavin (TF1), theaflavin-3-monogallate (TF2A), theaflavin-30-monogallate (TF2B), and theaflavin-3,30-digallate (TF3) (Fig. 14), have a considerable inhibitory effect on the HSV-1 lytic cycle, with TF3 being the most effective. TF3 at the concentration of 50 μM and above was sufficient to inhibit >99% of HSV-1 viral particles production.222 The anti-HSV-1 effect of TF3 was confirmed to be due to its direct effect on the virions. TF3 did not show significant antiviral effect after the virus had already penetrated the cells and treating Vero or A549 test cells with TF3 for 1 h prior to infection or at different times post infection did not inhibit HSV-1 production, either. Furthermore, a major advantage of theaflavins is their low cytotoxicity and stability under non-favorable conditions such as acidic pH, high temperature and high humidity as well as low cost.
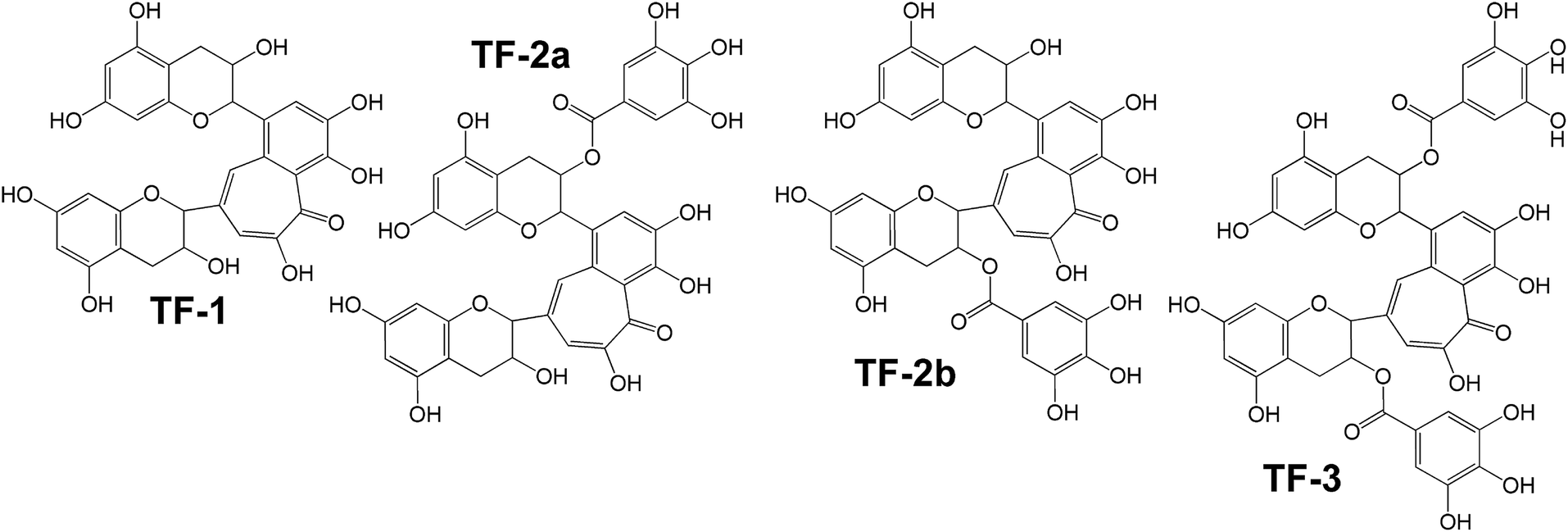 | ||
| Fig. 14 Structures of theaflavin polyphenols. | ||
5. Conclusions and future prospects
The present review indicates that despite high incidence of herpes virus (especially HSV-1) infections observed worldwide and extensive research on this virus, there has been only very limited progress in the development of novel drugs while the existing ones, mostly the DNA replication inhibitors (like ACV), have considerable limitations ranging from low bioavailability to viral resistance to these compounds. To avoid problems with the delivery the pro-drugs or novel drug delivery systems were developed. Growing attention has been also paid to the synthesis of novel non-nucleoside compounds and on using natural products as possible anti-HSV-1 agents. However, the progress in that research is not satisfactory. Still, there are no drugs or vaccines available which would allow eradication the virus, also because any candidate vaccine would have to be efficient against both HSV-1 and HSV-2 due to changing epidemiology of genital herpes.223,224 The HSV viral infection persists over the life span of the person causing not only the troublesome blisters on the skin but also, as the results of the recent studies indicated, blindness or neurodegenerative diseases. One of the reasons for that is still limited knowledge on specific interactions between various chemical compounds and cell membrane (especially at receptor sides) and with virus (especially viral envelope). There is a need to identify the compounds which would prevent the cell-to-cell spread of the virus. The viral infections could be limited if the mechanism of the interactions between cell membrane/viral envelope and novel, carefully designed and synthesized compounds, both synthetic or modified natural ones, was studied and understood.Acknowledgements
This work was supported by the National Science Centre, Poland in the form of the grant No. 2014/13/B/ST5/04510.References
- S. Boulant, M. Stanifer and P.-Y. Lozach, Viruses, 2015, 7, 2794 CrossRef CAS PubMed.
- U. F. Greber, Cell. Mol. Life Sci., 2002, 59, 608 CrossRef CAS PubMed.
- J. Mercer, M. Schelhaas and A. Helenius, Annu. Rev. Biochem., 2010, 79, 803 CrossRef CAS PubMed.
- A. Arvin, G. Campadelli-Fiume, E. Mocarski, P. S. Moore, B. Roizman, R. Whitley and K. Yamanishi, Human Herpesviruses. Biology, Therapy, and Immunoprophylaxis, Cambridge University Press, Cambridge, 2007 Search PubMed.
- M. L. Cook, V. B. Bastone and J. G. Stevens, Infect. Immun., 1974, 9, 946 CAS.
- F. Xu, M. R. Sternberg, B. J. Kottiri, G. M. McQuillan, F. K. Lee, A. J. Nahmias, S. M. Berman and L. E. Markowitz, JAMA, 2006, 296, 964 CrossRef CAS PubMed.
- R. Rubicz, C. T. Leach, E. Kraig, N. V. Dhurandhar, R. Duggirala, J. Blangero, J. R. Yolken and H. H. Goring, Hum. Hered., 2011, 72, 133 CrossRef PubMed.
- M. Fatahzadeh and R. A. Schwartz, J. Am. Acad. Dermatol., 2007, 57, 737 CrossRef PubMed.
- J. S. Smith and N. J. Robinson, J. Infect. Dis., 2002, 186(suppl. 1), S3 CrossRef PubMed.
- J. G. Stevens, A. B. Nesburn and M. L. Cook, Nature, New Biol., 1972, 235, 216 CrossRef CAS.
- J. G. Stevens and M. L. Cook, Cancer Res., 1973, 33, 1399 CAS.
- S. L. Wechsler, A. B. Nesburn, R. Watson, S. M. Slanina and H. Ghiasi, J. Virol., 1988, 62, 4051 CAS.
- G. C. Perng, E. C. Dunkel, P. E. Geary, S. M. Slanina, H. Ghiasi, R. Kaiwar, A. B. Nesburn and S. L. Wechsler, J. Virol., 1994, 68, 8045 CAS.
- G. C. Perng, S. M. Slanina, A. Yukht, H. Ghiasi, A. B. Nesburn and S. L. Wechsler, J. Virol., 2000, 74, 1885 CrossRef CAS PubMed.
- M. L. Cook, V. B. Bastone and J. G. Stevens, Infect. Immun., 1974, 9, 946 CAS.
- J. C. Zwaagstra, H. Ghiasi, A. B. Nesburn and S. L. Wechsler, Virology, 1991, 182, 287 CrossRef CAS PubMed.
- R. Rautemaa, T. Helander and S. Meri, Immunology, 2002, 106, 404 CrossRef CAS PubMed.
- G. C. Perng and C. Jones, Interdiscip. Perspect. Infect. Dis., 2010, 262415 Search PubMed.
- E. S. Barton, D. W. White, J. S. Cathelyn, K. A. Brett-McClellan, M. Engle, M. S. Diamond, V. L. Miller and H. W. Virgin, Nature, 2007, 447, 326 CrossRef CAS PubMed.
- M. Ahmed and N. W. Fraser, J. Virol., 2001, 75, 12070 CrossRef CAS PubMed.
- L. J. Al-Dujaili, P. P. Clerkin, C. Clement, H. E. McFerrin, P. S. Bhattacharjee, E. D. Varnell, H. E. Kaufman and J. M. Hill, Future Microbiol., 2011, 6, 877 CrossRef PubMed.
- D. Shukla and P. G. Spear, J. Clin. Invest., 2001, 108, 503 CrossRef CAS PubMed.
- R. J. Whitley and B. Roizman, Lancet, 2001, 357, 1513 CrossRef CAS.
- T. J. Liesegang, Cornea, 2001, 20, 1 CrossRef CAS PubMed.
- T. J. Liesegang, L. J. Melton, P. J. Daly and D. M. Ilstrup, Arch. Ophthalmol., 1989, 107, 1155 CrossRef CAS PubMed.
- E. Miserocchi, G. Modorati, L. Galli and P. Rama, Am. J. Ophthalmol., 2007, 144, 547 CrossRef CAS PubMed.
- H. S. Toma, A. T. Murina, R. G. Areaux, D. M. Neumann, P. S. Bhattacharjee, T. P. Foster, H. E. Kaufman and J. M. Hill, Semin. Ophthalmol., 2008, 23, 249 CrossRef PubMed.
- B. F. Khan and D. Pavan-Langston, Int. Ophthalmol. Clin., 2004, 44, 103 CrossRef PubMed.
- R. F. Itzhaki, W. R. Lin, D. Shang, G. K. Wilcock, B. Faragher and G. A. Jamieson, Lancet, 1997, 349, 241 CrossRef CAS.
- L. Letenneur, K. Pérès, H. Fleury, I. Garrigue, P. Barberger-Gateau, C. Helmer, J. M. Orgogozo, S. Gauthier and J. F. Dartigues, PLoS One, 2008, 3, e3637 Search PubMed.
- M. A. Wozniak, A. L. Frost, C. M. Preston and R. F. Itzhaki, PLoS One, 2011, 6, e25152 CAS.
- R. Piacentini, G. de Chiara, D. D. Li Puma, C. Ripoli, M. E. Marcocci, E. Garaci, A. T. Palamara and C. Grassi, Front. Pharmacol., 2014, 5, 97 CrossRef PubMed.
- S. A. Harris and E. A. Harris, J. Alzheimer's Dis., 2015, 48, 319 CrossRef CAS PubMed.
- X. Jiang, D. Brown, N. Osorio, C. Hsiang, L. BenMohamed and S. L. Wechsler, J. NeuroVirol., 2015 DOI:10.1007/s13365-015-0362-y.
- R. L. Neve and F. Lim, Current Protocols in Neuroscience, 2012, 61, 1 Search PubMed.
- D. Kuruppu and K. K. Tanabe, Cancer Gene Ther., 2015, 22, 506 CrossRef CAS PubMed.
- D. Furlong, H. Swift and B. Roizman, J. Virol., 1972, 10, 1071 CAS.
- D. H. Russell, The structural and metabolic involvment of polyamines with herpes simplex virus, in Polyamines in normal and neoplastic growth, Raven Press, New York, 1973 Search PubMed.
- K. Borchers, M. Goltz and H. Ludwig, Acta Vet. Hung., 1994, 42, 217 CAS.
- D. J. McGeoch, J. Cell Sci., Suppl., 1987, 7, 67 CrossRef CAS.
- I. Liashkovich, W. Hafezi, J. E. Kühn, H. Oberleithner, A. Kramer and V. Shahin, J. Cell Sci., 2008, 121, 2287 CrossRef CAS PubMed.
- R. J. Geraghty, C. Krummenacher, G. H. Cohen, R. J. Eisenberg and P. G. Spear, Science, 1998, 5369, 1618 CrossRef.
- C. Krummenacher, A. V. Nicola, J. C. Whitbeck, H. Lou, W. Hou, J. D. Lambris, R. J. Geraghty, P. G. Spear, G. H. Cohen and R. J. Eisenberg, J. Virol., 1998, 72, 7064 CAS.
- R. I. Montgomery, M. S. Warner, B. J. Lum and P. G. Spear, Cell, 1996, 87, 427 CrossRef CAS PubMed.
- D. Shukla, J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg and P. G. Spear, Cell, 1999, 99, 13 CrossRef CAS PubMed.
- J. C. Whitbeck, C. Peng, H. Lou, R. Xu, S. H. Willis, M. Ponce de Leon, T. Peng, A. V. Nicola, R. I. Montgomery, M. S. Warner, A. M. Soulika, L. A. Spruce, W. T. Moore, J. D. Lambris, P. G. Spear, G. H. Cohen and R. J. Eisenberg, J. Virol., 1997, 71, 6083 CAS.
- F. Cocchi, M. Lopez, L. Menotti, M. Aoubala, P. Dubreuil and G. Campadelli-Fiume, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15700 CrossRef CAS.
- G. Lu, N. Zhang, J. Qi, Y. Li, Z. Chen, C. Zheng, G. F. Gao and J. Yan, J. Virol., 2014, 88, 13678 CrossRef CAS PubMed.
- C. Pasero, D. E. Speiser, L. Derré and D. Olive, Curr. Opin. Pharmacol., 2012, 12, 478 CrossRef CAS PubMed.
- A. H. Karaba, S. J. Kopp and R. Longnecker, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20649 CrossRef CAS PubMed.
- V. Tiwari, C. Clement, M. B. Duncan, J. Chen, J. Liu and D. Shukla, J. Gen. Virol., 2004, 85, 805 CrossRef CAS PubMed.
- V. Tiwari, C. Clement, D. Xu, T. Valyi-Nagy, B. Y. Yue, J. Liu and D. Shukla, J. Virol., 2006, 80, 8970 CrossRef CAS PubMed.
- E. E. Heldwein, H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg and S. C. Harrison, Science, 2006, 5784, 217 CrossRef PubMed.
- P. G. Spear, Cell. Microbiol., 2004, 6, 401 CrossRef CAS PubMed.
- E. Avitabile, C. Forghieri and G. Campadelli-Fiume, J. Virol., 2007, 81, 11532 CrossRef CAS PubMed.
- J. C. Whitbeck, Y. Zuo, R. S. Milne, G. H. Cohen and R. J. Eisenberg, J. Virol., 2006, 80, 3773 CrossRef CAS PubMed.
- A. V. Nicola, A. M. McEvoy and S. E. Straus, J. Virol., 2003, 77, 5324 CrossRef CAS PubMed.
- K. Radtke, D. Kieneke, A. Wolfstein, K. Michael, W. Steffen, T. Scholz, A. Karger and B. Sodeik, PLoS Pathog., 2010, 7, e1000991 Search PubMed.
- M. McElwee, F. Beilstein, M. Labetoulle, F. J. Rixon and D. Pasdeloup, J. Virol., 2013, 87, 11008 CrossRef CAS PubMed.
- C. Morgan, H. M. Rose and B. Mednis, J. Virol., 1968, 5, 507 Search PubMed.
- K. Miyamoto and C. Morgan, J. Virol., 1971, 8, 910 CAS.
- P. M. Ojala, B. Sodeik, M. W. Ebersold, U. Kutay and A. Helenius, Mol. Cell. Biol., 2000, 20, 4922 CrossRef CAS PubMed.
- B. S. Albright, J. Nellissery, R. Szczepaniak and S. K. Weller, J. Virol., 2011, 85, 8616 CrossRef CAS PubMed.
- H. Hofemeister and P. O'Hare, J. Virol., 2008, 82, 8392 CrossRef CAS PubMed.
- H. G. Page and G. S. Read, J. Virol., 2010, 84, 6886 CrossRef CAS PubMed.
- T. Gerster and R. G. Roeder, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6347 CrossRef CAS.
- T. M. Kristie, J. H. LeBowitz and P. A. Sharp, EMBO J., 1989, 13, 4229 Search PubMed.
- I. Muylaert and P. Elias, J. Biol. Chem., 2007, 282, 10865 CrossRef CAS PubMed.
- J. A. Dembowski and N. A. DeLuca, PLoS Pathog., 2015, 11, e1004939 Search PubMed.
- R. Hagglund and B. Roizman, J. Virol., 2004, 78, 2169 CrossRef CAS PubMed.
- H. Gu, Y. Liang, G. Mandel and B. Roizman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7571 CrossRef CAS PubMed.
- C. M. Preston, J. Virol., 1979, 29, 275 CAS.
- R. J. Watson and J. B. Clements, Nature, 1980, 5763, 329 CrossRef.
- F. McGregor, A. Phelan, J. Dunlop and J. B. Clements, J. Virol., 1996, 70, 1931 CAS.
- L. A. Samaniego, A. L. Webb and N. A. DeLuca, J. Virol., 1995, 69, 5705 CAS.
- S. L. Uprichard and D. M. Knipe, J. Virol., 1996, 70, 1969 CAS.
- P. J. Godowski and D. M. Knipe, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 256 CrossRef CAS.
- M. Olesky, E. E. McNamee, C. Zhou, T. J. Taylor and D. M. Knipe, Virology, 2005, 331, 94 CrossRef CAS PubMed.
- M. C. Long, V. Leong, P. A. Schaffer, C. A. Spencer and S. A. Rice, J. Virol., 1999, 73, 5593 CAS.
- S. J. Advani, R. Brandimarti, R. R. Weichselbaum and B. Roizman, J. Virol., 2000, 74, 8 CrossRef CAS PubMed.
- L. Chang, W. J. Godinez, I. H. Kim, M. Tektonidis, P. de Lanerolle, R. Eils, K. Rohr and D. M. Knipe, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E136 CrossRef PubMed.
- L. Song, M. Chaudhuri, C. W. Knopf and D. S. Parris, J. Biol. Chem., 2004, 279, 18535 CrossRef CAS PubMed.
- P. E. Boehmer and I. R. Lehman, Annu. Rev. Biochem., 1997, 66, 347 CrossRef CAS PubMed.
- Y. Chen, P. Bai, S. Mackay, G. Korza, J. H. Carson, R. D. Kuchta and S. K. Weller, J. Virol., 2011, 85, 968 CrossRef CAS PubMed.
- S. Chattopadhyay, Y. Chen and S. K. Weller, Front. Biosci., 2006, 11, 2213 CrossRef CAS.
- W. W. Newcomb, F. L. Homa and J. C. Brown, J. Virol., 2006, 80, 6286 CrossRef CAS PubMed.
- W. W. Newcomb, D. R. Thomsen, F. L. Homa and J. C. Brown, J. Virol., 2003, 77, 9862 CrossRef CAS PubMed.
- W. W. Newcomb, F. L. Homa, D. R. Thomsen, F. P. Booy, B. L. Trus, A. C. Steven, J. V. Spencer and J. C. Brown, J. Mol. Biol., 1996, 263, 432 CrossRef CAS PubMed.
- X. Zhang, S. Efstathiou and A. Simmons, Virology, 1994, 202, 530 CrossRef CAS PubMed.
- W. W. Newcomb, R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller and J. C. Brown, J. Virol., 2001, 75, 10923 CrossRef CAS PubMed.
- T. C. Mettenleiter, F. Müller, H. Granzow and B. G. Klupp, Cell. Microbiol., 2013, 15, 170 CrossRef CAS PubMed.
- L. A. Morrison and G. S. DeLassus, Nucleus, 2011, 4, 271 CrossRef PubMed.
- D. C. Johnson and J. D. Baines, Nat. Rev. Microbiol., 2011, 9, 382 CrossRef CAS PubMed.
- J. N. Skepper, A. Whiteley, H. Browne and A. Minson, J. Virol., 2001, 75, 5697 CrossRef CAS PubMed.
- G. Campadelli, R. Brandimarti, C. Di Lazzaro, P. L. Ward, B. Roizman and M. R. Torrisi, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 2798 CrossRef CAS.
- T. W. Wisner and D. C. Johnson, J. Virol., 2004, 78, 11519 CrossRef CAS PubMed.
- M. P. Nicoll, J. T. Proença and S. Efstathiou, FEMS Microbiol. Rev., 2012, 36, 684 CrossRef CAS PubMed.
- C. Ramakrishna, A. Ferraioli, A. Calle, T. K. Nguyen, H. Openshaw, P. S. Lundberg, P. Lomonte and E. M. Cantin, PLoS Pathog., 2015, 11, e1004730 Search PubMed.
- Y. Imai, K. Apakupakul, P. R. Krause, W. P. Halford and T. P. Margolis, J. Virol., 2009, 83, 7873 CrossRef CAS PubMed.
- M. Inman, G. C. Perng, G. Henderson, H. Ghiasi, A. B. Nesburn, S. L. Wechsler and C. Jones, J. Virol., 2001, 75, 3636 CrossRef CAS PubMed.
- O. Akhova, M. Bainbridge and V. Misra, J. Virol., 2005, 79, 14708 CrossRef CAS PubMed.
- G. Zhou, T. Du and B. Roizman, Viruses, 2013, 5, 1208 CrossRef CAS PubMed.
- M. L. Nogueira, V. E. Wang, D. Tantin, P. A. Sharp and T. M. Kristie, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1473 CrossRef CAS PubMed.
- B. Roizman, G. Zhou and T. Du, J. NeuroVirol., 2011, 17, 512 CrossRef PubMed.
- R. D. Everett, Methods Mol. Biol., 2014, 1144, 1 Search PubMed.
- A. Thessicar, P. J. Park and D. Shukla, Rev. Med. Virol., 2013, 23, 194 CrossRef PubMed.
- J. P. Engel, J. A. Englund, C. V. Fletcher and E. L. Hill, JAMA, 1990, 263, 1662 CrossRef CAS PubMed.
- R. J. Whitley, D. W. Kimberlin and B. Roizman, Clin. Infect. Dis., 1998, 26, 541 CrossRef CAS PubMed.
- J. E. Reardon and T. Spector, Adv. Pharmacol., 1991, 22, 1 CAS.
- D. Ramyadevi and K. S. Rajan, RSC Adv., 2015, 5, 12956 RSC.
- M. Tod, F. Lokiec, R. Bidault, F. Debony, O. Petitjean and Y. Aujard, Antimicrob. Agents Chemother., 2001, 45, 150 CrossRef CAS PubMed.
- K. R. Beutner, Antiviral Res., 1995, 28, 281 CrossRef CAS PubMed.
- D. Ormrod, L. J. Scott and C. M. Perry, Drugs, 2000, 59, 839 CrossRef CAS PubMed.
- G. E. Granero and G. L. Amidon, Int. J. Pharm., 2006, 317, 14 CrossRef CAS PubMed.
- S. Weller, M. R. Blum, M. Doucette, T. Burnette, D. M. Cederberg, P. de Miranda and M. L. Smiley, Clin. Pharmacol. Ther., 1993, 54, 595 CrossRef CAS PubMed.
- P. J. Sinko and P. V. Balimane, Biopharm. Drug Dispos., 1998, 9, 209 CrossRef.
- C. M. Perry and A. J. Wagstaff, Drugs, 1995, 50, 396 CrossRef CAS PubMed.
- H. Degreef, L. Andrejevic, F. Aoki, J. Arend, R. Ashton, W. Debacker, K. Bartlett, W. B. Vanblokland, S. Bishop and R. Boon, et al., Int. J. Antimicrob. Agents, 1994, 4, 241 CrossRef CAS PubMed.
- T. Matthews and R. Boehme, Rev. Infect. Dis., 1988, 10(suppl. 3), S490 CrossRef CAS PubMed.
- N. Castela, N. Vermerie, F. Chast, H. Sauvageon-Martre, J. Denis, V. Godard, P. Goldschmidt and Y. Pouliquen, J. Ocul. Pharmacol., 1994, 10, 439 CrossRef CAS PubMed.
- H. Shiota, T. Naito and Y. Mimura, Curr. Eye Res., 1987, 6, 241 CrossRef CAS PubMed.
- H. B. Hoh, C. Hurley, C. Claoue, M. Viswalingham, D. L. Easty, P. Goldschmidt and L. M. Collum, Br. J. Ophthalmol., 1996, 80, 140 CrossRef CAS PubMed.
- C. S. Crumpacker, Am. J. Med., 1992, 92, 3S CrossRef CAS PubMed.
- C. Danve-Szatanek, M. Aymard, D. Thouvenot, F. Morfin, G. Agius, I. Bertin, S. Billaudel, B. Chanzy, M. Coste-Burel and L. Finkielsztejn, et al., J. Clin. Microbiol., 2004, 42, 242 CrossRef CAS PubMed.
- S. Safrin, C. Crumpacker, P. Chatis, R. Davis, R. Hafner, J. Rush, H. A. Kessler, B. Landry and J. A. Mills, N. Engl. J. Med., 1991, 325, 551 CrossRef CAS PubMed.
- X. Xiong, J. L. Smith and M. S. Chen, Antimicrob. Agents Chemother., 1997, 41, 594 CAS.
- B. S. Anand, J. M. Hill, S. Dey, K. Maruyama, P. S. Bhattacharjee, M. E. Myles, Y. E. Nashed and A. K. Mitra, Invest. Ophthalmol. Visual Sci., 2003, 44, 2529 Search PubMed.
- K. Suresh, Z. Xiadong, T. S. Ravi and A. K. Mitra, Ophthalmol. Eye Dis., 2010, 2, 43 CAS.
- C. von Plessing Rossel, J. S. Carreño, M. Rodríguez-Baeza and J. B. Alderete, Quim. Nova, 2000, 23, 749 CrossRef CAS.
- B. Anroop, M. A. Nair, E. Bandar, J. W. Al-Dhubiab, H. Sree and A. Mueen, Drug Delivery, 2014, 7, 540 Search PubMed.
- M. Bencini, E. Ranucci, P. Ferruti, F. Trotta, M. Donalisio, M. Cornaglia, D. Lembo and R. Cavalli, J. Controlled Release, 2008, 126, 17 CrossRef CAS PubMed.
- R. Cavalli, M. Donalisio, A. Civra, P. Ferruti, E. Ranucci, F. Trotta and D. Lembo, J. Controlled Release, 2009, 137, 116 CrossRef CAS PubMed.
- D. Lembo, S. Swaminathan, M. Donalisio, A. Civra, L. Pastero, D. Aquilano, P. Vavia, F. Trotta and R. Cavalli, Int. J. Pharm., 2013, 443, 262 CrossRef CAS PubMed.
- S. Shahsavari, E. V. Farahani, M. Ardjmand and F. A. Dorkoosh, Curr. Nanosci., 2014, 10, 521 CrossRef CAS.
- A. Gandhi, S. Jana and K. K. Sen, Int. J. Biol. Macromol., 2014, 67, 478 CrossRef CAS PubMed.
- M. M. Al-Subaie, K. M. Hosny, K. M. El-Say, T. Aahmed and M. A. Bader, Int. J. Nanomed., 2015, 10, 3973 Search PubMed.
- Y. S. R. Krishnaiah, X. Xu, Z. Rahman, Y. Yang, U. Katragadda, R. Lionberger, J. R. Peters, K. Uhl and M. A. Khan, Int. J. Pharm., 2014, 475, 110 CrossRef CAS PubMed.
- B. Chaudhary and S. Verma, Sci. World J., 2014, 2014, 280928 Search PubMed.
- D. Ramyadevi and S. R. Kalpoondi, RSC Adv., 2015, 5, 12956 RSC.
- P. Chetoni, S. Rossi, S. Burgalassi, D. Monti, S. Mariotti and M. F. Saettone, J. Ocul. Pharmacol. Ther., 2004, 20, 169 CrossRef CAS PubMed.
- G. S. El-Fekya, G. Zayedb and A. R. H. Farrag, Int. J. Pharm. Pharm. Sci., 2013, 5, 213 Search PubMed.
- D. Gulsen and A. Chauhan, Invest. Ophthalmol. Visual Sci., 2004, 45, 2342 Search PubMed.
- B. Bareiss, M. Ghorbani, F. Li, J. A. Blake, J. C. Scaiano, J. Zhang, C. Deng, K. Merrett, J. L. Harden, F. Diaz-Mitoma and M. Griffith, Open Tissue Eng. Regener. Med. J., 2010, 3, 10 CrossRef CAS.
- S. Burrel, C. Aime, L. Hermet, Z. Ait-Arkoub, H. Agut and D. Boutolleau, Antiviral Res., 2013, 100, 365 CrossRef CAS PubMed.
- R. Stránská, R. Schuurman, E. Nienhuis, I. W. Goedegebuure, M. Polman, J. F. Weel, P. M. Wertheim-Van Dillen, R. J. Berkhout and A. M. van Loon, J. Clin. Virol., 2005, 32, 7 CrossRef PubMed.
- D. Malvy, M. Treilhaud, S. Bouée, A. Crochard, D. Vallée, A. El Hasnaoui, M. Aymard and the RESSAC Study Group, Clin. Infect. Dis., 2005, 41, 320 CrossRef CAS PubMed.
- H. Izzedine, V. Launay-Vacher and G. Deray, Am. J. Kidney Dis., 2005, 45, 804 CrossRef CAS PubMed.
- S. Karampuri, D. Ojha, P. Bag, H. Chakravarty, C. Bal, D. Chattopadhyay and A. Sharon, RSC Adv., 2014, 4, 17354 RSC.
- K. Pegklidou, N. Papastavrou, P. Gkizis, D. Komiotis, J. Balzarini and I. Nicolaou, Med. Chem., 2015, 11, 602 CrossRef CAS.
- M. E. Dávola, G. I. Mazaira, M. D. Galigniana, L. E. Alché, J. A. Ramírez and A. A. Barquero, Antiviral Res., 2015, 122, 55 CrossRef PubMed.
- V. Castilla, J. A. Ramírez and C. E. Coto, Curr. Med. Chem., 2010, 17, 1858 CrossRef CAS PubMed.
- A. V. Nicola, J. Hou, E. O. Major and E. S. Straus, J. Virol., 2005, 79, 7609 CrossRef CAS PubMed.
- L. McClain, Y. Z. Hoyee Cheng, A. Ghosh, P. Piazza, M. B. Yeee, S. Kumar, J. Milosevic, D. C. Bloomi, R. Arav-Boger, P. R. Kinchingtonf, R. Yolkenj, V. Nimgaonkar and L. D'Aiuto, Antiviral Res., 2015, 121, 16 CrossRef CAS PubMed.
- C. A. Harley, A. Dasgupta and D. W. Wilson, J. Virol., 2001, 75, 1236 CrossRef CAS PubMed.
- T. J. Nieland, Y. Feng, J. X. Brown, T. D. Chuang, P. D. Buckett, J. Wang, X. S. Xie, T. E. McGraw, T. Kirchhausen and M. Wessling-Resnick, Traffic, 2004, 5, 478 CrossRef CAS PubMed.
- J. A. Wojcechowskyj and R. W. Doms, Viruses, 2010, 2, 1106 CrossRef PubMed.
- L. E. Pope, J. F. Marcelletti, L. R. Katz, J. Y. Lin, D. H. Katz, M. L. Parish and P. G. Spear, Antiviral Res., 1998, 40, 85 CrossRef CAS PubMed.
- D. H. Katz, J. F. Marcelletti, M. H. Khalil, L. E. Pope and L. R. Katz, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10825 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef.
- V. Pirrone, B. Wigdahl and F. C. Krebs, Antiviral Res., 2011, 90, 168 CrossRef CAS PubMed.
- M. Donalisio, E. Ranucci, V. Cagno, A. Civra, A. Manfredi, R. Cavalli, P. Ferruti and D. Lembo, Antimicrob. Agents Chemother., 2014, 58, 6315 CrossRef PubMed.
- R. V. Alasino, S. F. Ausar, I. D. Bianco, L. Castagna, M. Contigiani and D. M. Beltramo, Macromol. Biosci., 2005, 5, 207 CrossRef CAS PubMed.
- R. V. Alasino, I. D. Bianco, M. S. Vitali, J. A. Zarzur and D. M. Beltramo, Macromol. Biosci., 2007, 7, 1132 CrossRef CAS PubMed.
- L. Zeitlin, K. J. Whaley, T. A. Hegarty, T. R. Moench and R. A. Cone, Contraception, 1997, 56, 329 CrossRef CAS PubMed.
- R. A. Anderson, K. Feathergill, X. Diao, M. Cooper, R. Kirkpatrick, P. Spear, D. P. Waller, C. Chany, G. F. Doncel, B. Herold and L. J. Zaneveld, J. Androl., 2000, 21, 862 CAS.
- M. Qiu, Y. Chen, S. Song, H. Song, Y. Chu, Z. Yuan, L. Cheng, D. Zheng, Z. Chen and Z. Wu, Antiviral Res., 2012, 96, 138 CrossRef CAS PubMed.
- Y. Okada and J. Kim, Virology, 1972, 50, 507 CrossRef CAS PubMed.
- M. Ito and A. L. Barron, J. Virol., 1974, 13, 1312 CAS.
- S. Galdiero, M. Vitiello, M. D'Isanto, A. Falanga, M. Cantisani, H. Browne, C. Pedone and M. Galdiero, ChemBioChem, 2008, 9, 758 CrossRef CAS PubMed.
- R. Akkarawongsa, N. E. Pocaro, G. Case, A. W. Kolb and C. R. Brandt, Antimicrob. Agents Chemother., 2009, 53, 987 CrossRef CAS PubMed.
- B. Shogan, L. Kruse, G. B. Mulamba, A. Hu and D. M. Coen, J. Virol., 2006, 80, 4740 CrossRef CAS PubMed.
- R. D. Dix, L. Pereira and J. R. Baringer, Infect. Immun., 1981, 34, 192 CAS.
- M.-J. Oh, J. Akhtar, P. Desai and D. Shukla, Biochem. Biophys. Res. Commun., 2010, 391, 176 CrossRef CAS PubMed.
- J. Akhtar and D. Shukla, FEBS J., 2009, 276, 7228 CrossRef CAS PubMed.
- W. Cai, B. Gu and S. Person, J. Virol., 1998, 62, 2596 Search PubMed.
- W. Cai, S. Person, C. DebRoy and B. Gu, J. Mol. Biol., 1988, 201, 575 CrossRef CAS PubMed.
- W. Cai, S. Person, S. C. Warner, J. Zhou and N. A. DeLuca, J. Virol., 1987, 61, 714 CAS.
- H.-Y. Chang, G. H. Cohen and R. J. Eisenberg, J. Virol., 1994, 68, 2529 Search PubMed.
- V. Tiwaria, S. Y. Shukla and D. Shukla, Antiviral Res., 2009, 84, 67 CrossRef PubMed.
- A. J. Bolmstedt, B. R. O'Keefe, S. R. Shenoy, J. B. McMahon and M. R. Boyd, Mol. Pharmacol., 2001, 59, 949 CAS.
- S. R. Shenoy, B. R. O'Keefe, A. J. Bolmstedt, L. K. Cartner and M. R. Boyd, J. Pharmacol. Exp. Ther., 2001, 297, 704 CAS.
- V. Tiwari, S. Y. Shukla and D. A. Shukla, Antiviral Res., 2009, 84, 67 CrossRef CAS PubMed.
- C. C. Tsai, P. Emau, Y. Jiang, M. B. Agy, R. J. Shattock, A. Schmidt, W. R. Morton, K. R. Gustafson and M. R. Boyd, AIDS Res. Hum. Retroviruses, 2004, 20, 11 CrossRef CAS PubMed.
- A. Luganini, S. F. Nicoletto, L. Pizzuto, G. Pirri, A. Giuliani, S. Landolfo and G. Gribaudo, Antimicrob. Agents Chemother., 2011, 55, 3231 CrossRef CAS PubMed.
- P. Dogra, E. B. Martin, A. Williams, R. L. Richardson, J. S. Foster, N. Hackenback, S. J. Kennel, T. E. Sparer and J. S. Wall, PLoS One, 2015, 10, e0126239 Search PubMed.
- S. Ray, C. A. Pujol, E. B. Damonte and B. Ray, Carbohydr. Polym., 2015, 131, 315 CrossRef CAS PubMed.
- E. A. Harden, R. Falshaw, S. M. Carnachan, E. R. Kern and M. N. Prichard, Antiviral Res., 2009, 83, 282 CrossRef CAS PubMed.
- A. J. Nahamias and S. Kibrick, J. Bacteriol., 1964, 87, 1060 Search PubMed.
- A. Vaheri, Acta Pathol. Microbiol. Scand., 1964, 171, 1 Search PubMed.
- A. P. Dyer, B. W. Banfield, D. Martindale, D. M. Spannier and F. Tufaro, J. Virol., 1997, 71, 191 CAS.
- R. A. Anderson, K. A. Feathergill, X. H. Diao, M. D. Cooper, R. Kirkpatrick, B. C. Herold, G. F. Doncel, C. J. Chany, D. P. Waller, W. F. Rencher and L. J. Zaneveld, J. Androl., 2002, 23, 426 CAS.
- V. L. Campos, D. F. Kawano, D. B. da Silva Jr and I. Carvalho, Carbohydr. Polym., 2009, 77, 167 CrossRef.
- E. B. Damonte, M. C. Matulewicz and A. S. Cerezo, Curr. Med. Chem., 2004, 11, 2399 CrossRef CAS PubMed.
- A. A. Kolender, C. A. Pujol, E. B. Damonte, A. S. Cerezo and M. C. Matulewicz, An. Asoc. Quim. Argent., 1998, 86, 304 Search PubMed.
- I. Wijesekara, R. Pangestuti and S.-K. Kim, Carbohydr. Polym., 2011, 84, 14 CrossRef CAS.
- A. M. de Godoi, L. C. Faccin-Galhardi, N. Lopes, C. Nozawa, R. R. de Almeida, N. M. P. S. Ricardo and R. E. C. Linhares, Curr. Pharm. Biotechnol., 2015, 16, 1024 CAS.
- V. A. Cosenza, D. A. Navarro, C. A. Pujol, E. B. Damonte and C. A. Stortz, Carbohydr. Polym., 2015, 128, 199 CrossRef CAS PubMed.
- M.-G. Zhong, Y.-F. Xiang, X.-X. Qiu, Z. Liu, K. Kitazato and Y.-F. Wang, RSC Adv., 2013, 3, 313 RSC.
- L. T. Lin, T. Y. Chen, C. Y. Chung, R. S. Noyce, T. B. Grindley, C. McCormick, T. C. Lin, G. H. Wang, C. C. Lin and C. D. Richardson, J. Virol., 2011, 85, 4386 CrossRef CAS PubMed.
- R. J. Sydiskis, D. G. Owen, J. L. Lohr, K. H. Rosler and R. N. Blomster, Antimicrob. Agents Chemother., 1991, 35, 2463 CrossRef CAS PubMed.
- L. C. Lin, Y. C. Kuo and C. J. Chou, Planta Med., 2000, 66, 333 CrossRef CAS PubMed.
- G. Ferrea, A. Canessa, F. Sampietro, M. Cruciani, G. Romussi and D. Bassetti, Antiviral Res., 1993, 21, 317 CrossRef CAS PubMed.
- M. Arisawa, A. Fujita, T. Hayashi, K. Hayashi, H. Ochiai and N. Morita, Chem. Pharm. Bull., 1990, 38, 1624 CrossRef CAS PubMed.
- M. Marchetti, S. Piani, V. Pietropaolo, L. Seganti, R. Nicoletti and N. Orsi, J. Chemother., 1994, 7, 90 CrossRef PubMed.
- C. M. O. Simones, M. Amoros and L. Girre, Phytother. Res., 1999, 21, 317 Search PubMed.
- M. Baba, R. Snoeck, R. Pauwels and E. de Clercq, Antimicrob. Agents Chemother., 1988, 32, 1742 CrossRef CAS PubMed.
- M. Witvrouw, J. Desmyter and E. de Clercq, Antiviral Chem. Chemother., 1994, 5, 345 CrossRef.
- L. B. Lee, K. Hayashi, T. Hayashi, U. Sankawa and M. Maeda, Planta Med., 1999, 65, 439 CrossRef PubMed.
- J. Neyts, R. Snoeck, D. Schols, J. Balzarini, J. D. Esko, A. VanSchepdael and E. de Clercq, Virology, 1992, 189, 48 CrossRef CAS PubMed.
- P. P. Jagodzinski, R. Wiaderkiewicz, G. Kurzawski, M. Kloczewiak, H. Nakashima, E. Hyjek, N. Yamamoto, T. Uryu, Y. Kaneko, M. P. Posner and D. Kozbor, Virology, 1994, 202, 735 CrossRef CAS PubMed.
- W. Zhu, V. E. C. Ooi, P. K. S. Chan and P. O. Ang Jr, Biochem. Cell Biol., 2003, 81, 25 CrossRef CAS PubMed.
- W. Zhua, L. C. M. Chiu, V. E. C. Ooi, P. K. S. Chan and P. O. Ang Jr, Phytomedicine, 2006, 13, 695 CrossRef PubMed.
- M. Kim, J. H. Yim, S.-Y. Kim, H. S. Kim, W. G. Lee, S. J. Kim, P.-S. Kang and C.-K. Lee, Antiviral Res., 2012, 93, 253 CrossRef CAS PubMed.
- W. Wang, S.-X. Wang and H.-S. Guan, Mar. Drugs, 2012, 10, 2795 CrossRef PubMed.
- D. F. Argenta, I. T. Silva, V. L. Bassani1, L. S. Koester, H. F. Teixeira and C. M. O. Simoes, Arch. Virol., 2015, 160, 2335 CrossRef CAS PubMed.
- R. R. Hafidh, A. S. Abdulamir, F. A. Bakar, Z. Sekawi, F. Jahansheri and F. A. Jalilian, BMC Complementary Altern. Med., 2015, 15, 179 CrossRef PubMed.
- Á. L. Álvarez, S. Habtemariam, A. E. Abdel Moneim, S. Melón, K. P. Dalton and F. Parra, Antiviral Res., 2015, 119, 8 CrossRef PubMed.
- Y. Gong, K. M. Raj, C. A. Luscombe, I. Gadawski, T. Tam, J. Chu, D. Gibson, R. Carlson and S. L. Sacks, Antiviral Res., 2004, 64, 127 CrossRef CAS PubMed.
- J. Visalli, H. Ziobrowski, K. R. Badri, J. J. He, X. Zhang, S. R. Arumugam and H. Zhao, Bioorg. Med. Chem. Lett., 2015, 25, 3168 CrossRef PubMed.
- P. Bag, D. Ojha, H. Mukherjee, U. C. Halder, S. Mondal, A. Biswas, A. Sharon, L. van Kaer, S. Chakrabarty, G. Das, D. Mitra and D. Chattopadhyay, Antiviral Res., 2014, 105, 126 CrossRef CAS PubMed.
- D. Chen, A. Su, Y. Fu, X. Wang, X. Lv, W. Xu, S. Xu, H. Wang and Z. Wu, Antiviral Res., 2015, 123, 27 CrossRef CAS PubMed.
- A. de Oliveira, D. Prince, C.-Y. Lo, L. H. Lee and T.-C. Chu, Antiviral Res., 2015, 118, 56 CrossRef CAS PubMed.
- T. Tran, J. D. Druce, M. C. Catton, H. Kelly and C. J. Birch, Sex. Transm. Infect., 2004, 80, 277 CrossRef CAS PubMed.
- R. B. Belshe, P. A. Leone, D. I. Bernstein, A. Wald, M. J. Levin, J. T. Stapleton and I. Gorfinkel, et al., N. Engl. J. Med., 2012, 366, 34 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |