
Controlled growth of silver nanoparticles on carbon fibers for reinforcement of both tensile and interfacial strength
Caifeng Wanga,
Jun Lia,
Shaofan Suna,
Xiaoyu Lib,
Guangshun Wua,
Yuwei Wangac,
Fei Xiea and
Yudong Huang*a
aSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin 150001, China. E-mail: ydhuang.hit1@aliyun.com; Fax: +86 451 86221048; Tel: +86 451 86414806
bCollege of Life Sciences, Northeast Forestry University, Harbin 150040, China
cCollege of Materials Science and Engineering, Qiqihar University, Qiqihar 161006, China
First published on 21st January 2016
Abstract
Metal nanoparticles are commonly used for surface modification in fiber reinforced polymer composites because of their large specific surface area and electronic, magnetic and other related properties. In this study, morphology-controllable silver nanoparticles (Ag NPs) were deposited on a carbon fiber surface via a facile and green electro-chemical deposition method in the presence of poly(vinylpyrrolidone) (PVP). It was found that the presence of PVP and its molar ratio (in terms of repeating unit) relative to silver nitrate both played important roles in determining the geometric shape and size of the Ag NPs. Interestingly, electro-chemical deposition of Ag NPs improved both the tensile strength of the carbon single fiber and the interfacial property of the carbon fiber/epoxy composite by as much as 57.2% and 27.2%, respectively. Moreover, the Ag NPs-loaded carbon fibers exhibited superior electrical conductivity, which was a 2-fold enhancement as compared with that of the virgin carbon fibers. It meant that the Ag NPs-loaded carbon fibers could be used as ideal reinforcement materials for advanced aerospace systems.
1. Introduction
In the past two decades, carbon fiber reinforced polymer composites have been applied widely, ranging from aerospace and military to sport apparatus, owing to their high stiffness, high strength, light weight and excellent thermal resistance. It is well known that the interphase between fibers and the matrix is a crucial factor in the mechanical properties of carbon fiber composites.1–3 Therefore, researchers have proposed methods, such as electro-chemical deposition, oxidation treatment, plasma treatment and high energy irradiation, to treat the carbon fibers in order to improve the interfacial properties of the composites.4–7Owing to their large specific surface area, unique electronic and magnetic properties, nanoparticles have received more and more attention in the research of fiber reinforced polymer composites.8,9 It has been proved that the interfacial strength of nanoparticle modified fibers reinforced composites is improved. In recent years, some efforts to increase the surface energy (or wettability) and the surface roughness of carbon fibers have been made by modifying carbon fibers with nano-materials, such as carbon nanotubes (CNTs), graphene or metal nanoparticles.10–12 In our previous study, we tried several methods to improve the interfacial properties of fiber reinforced polymer composites, such as grafting POSS, CNT, graphene and metal-plating on carbon fiber.1,2,13,14 Among them, the metal-plating method shows many advantages, such as good conductivity, high strength and a uniform plating layer.15–20 Especially, Ag NPs seem to be the most popular metal among the highest electrical and thermal conductivity metals. It is believed that the control of the size, shape and structure of metal nanoparticles is technologically important because of the strong correlation between these parameters and the electrical and mechanical properties.21
Many methods have been proposed to prepare silver materials with different morphologies, such as hydrothermal method, the polyol process and electroless plating. However, these methods show many disadvantages, such as nonuniformity, long reaction time, chemical pollution and inefficiency. What is more, the improvement of interfacial properties is often followed by a sacrifice of fiber tensile strength owing to the etching effect during the treatment.22,23 Recently, electro-chemical deposition has been developed and proved to be an economical and green process that has been successfully applied in deposition of phosphors or catalysis coatings.24,25 Since Reuss et al. first observed the electric-field-induced motion of clay particles in water in 1807, many advantages of the reduction of metal particles from aqueous solutions have been found via the electro-chemical deposition method, such as high deposition rate, high throughput, good uniformity, controlled thickness, no need for binders and simplicity of scaling up.26,27 However, coating controlled Ag NPs on carbon fibers through electro-chemical deposition has not been reported yet. Furthermore, the relationship between surface morphology and mechanical properties of Ag NPs-loaded carbon fibers should be fully understood before use.
In this work, different morphologic structures of silver coatings (spherical and dendritic) have been fabricated on carbon fibers by electro-chemical deposition in aqueous solution. Ag NPs plated on the carbon fiber improved both the tensile strength of carbon single fiber and the interfacial properties of the composites. Moreover, the Ag NPs-loaded carbon fibers exhibited a superior electric conductivity compared with untreated carbon fibers. The relationship between surface morphology and the interfacial property of Ag NPs-loaded carbon fibers/resin composites was studied for the first time. This modified carbon fiber is expected to be used in fabricating conducting, shielding and wave-absorbing aerospace materials.
2. Materials and methods
2.1. Materials
Carbon fibers (T700SC-12000-50C, 12 K, tensile strength 4.9 GPa, diameter 7 μm, density 1.8 g cm−3) were purchased from Toray Industries, Inc. WSR618 epoxy resin (molecular weight 350–400) and methyl tetrahydrophthalic anhydride hardener were supplied by Sinopharm Chemical Reagent Co., Ltd. Silver nitrate (AgNO3) and PVP (molecular weight 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sigma-Aldrich. Deionized water (DI water, >18 MΩ cm) was used throughout the experiments. All chemicals were used as received unless stated otherwise.
000) were purchased from Sigma-Aldrich. Deionized water (DI water, >18 MΩ cm) was used throughout the experiments. All chemicals were used as received unless stated otherwise.
2.2. Experimental procedure
Carbon fibers were refluxed in acetone for 48 h to remove the polymer sizing and pollutants before used. Measured amounts of AgNO3 and PVP were dissolved in 140 ml of DI water and mixed well. The mixture was left unstirred for several minutes before used. Different PVP-to-silver ratios used in this work were listed in Table 1. An electro-chemical deposition system was established using AgNO3 and PVP aqueous as the support electrolyte, carbon fiber bundle as the working electrode (cathode) and copper as the counter electrode (anode), as shown in Fig. 1. The prepared samples and their deposition conditions are outlined in Table 1.| Sample | AgNO3 (mmol L−1) | PVP/AgNO3 molar ratios | Voltage (V cm−1) | Time (min) | |
|---|---|---|---|---|---|
| Sample 1 | CF/Ag-1 | 3 | 1.5 | 3 | 5 |
| Sample 2 | CF/Ag-2 | 3 | 5 | ||
| Sample 3 | CF/Ag-3 | 6 | 3 | 3 | 2 |
| Sample 4 | CF/Ag-4 | 3 | 5 | ||
| Sample 5 | CF/Ag-5 | 3 | 10 | ||
| Sample 6 | CF/Ag-6 | 0 | 5 | ||
| Sample 7 | CF/Ag-7 | 1.5 | 5 | ||
| Sample 8 | CF/Ag-8 | 30 | 1.5 | 3 | 5 |
| Sample 9 | CF/Ag-9 | 3 | 5 |
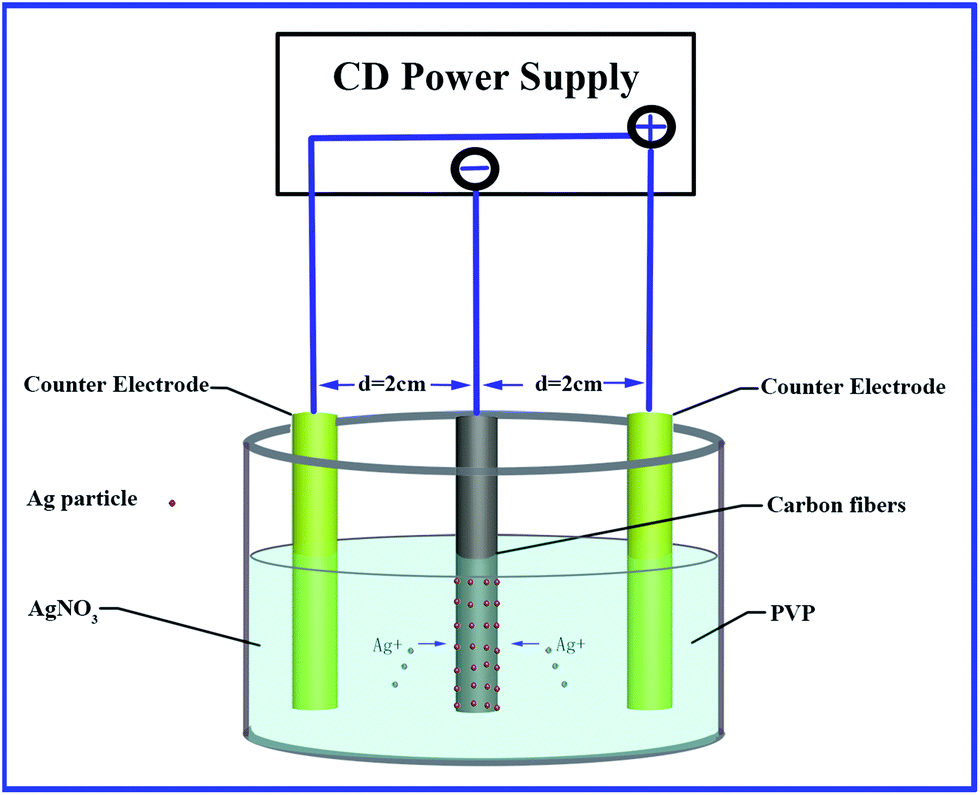 | ||
| Fig. 1 Schematic illustration of electro-chemical deposition of silver nanoparticles on carbon fiber. | ||
2.3. Characterization of the Ag NPs-loaded carbon fibers
The surface morphologies of the Ag NPs-loaded carbon fibers were observed by SEM (200FEG, Quanta FEI Inc. The USA) and all samples were sputtered with gold before doing SEM.The cross-section of silver coating was observed using TEM (Hitachi H-7650) at 200 kV. To prepare the TEM samples, the carbon fibers were moulded in a room-temperature curing epoxy resin and then the composite was cross sectioned into thin sections (<100 nm) using a microtome.
The surface roughness of the Ag NPs-loaded carbon fibers was characterized by AFM (Solver-P47H, NT-MDT, Russia), in which an area of 4 × 4 μm was scanned and evaluated by Rmax and Ra, respectively. Rmax stands for the maximum roughness, which is used for characterizing the differences between the maximum and minimum values of the height coordinates in the measuring range. Ra is the average roughness, representing the average value of roughness in the measuring range.
XPS (ESCALAB 220i-XL, VG, UK) was carried out to study the surface chemical constituents after electro-chemical deposition using a monochromatic Al Kα source (1486.6 eV) at a base pressure of 2 × 10−9 mbar. XPS Peak version 4.1 was used for data analysis.
The crystallographic structures of the materials were measured by a powder X-ray diffraction system (XRD, TTR-III) equipped with Cu K(α) radiation (λ = 0.15406 nm).
Single fiber tensile tests were performed on a universal testing machine (5569, Instron, USA) according to ASTMD3379-75. The results were analyzed with the Wellbull statistical method.
The electrical conductivity of the Ag NPs-loaded carbon fibers was measured using a two-point method (Keithley 2420 I-V, Keithley Instruments Inc.), as shown in Fig. 2(b) and (c). In order to immobilize the free fibers, the specimens were prepared in metal frames with length of approximately 40 mm (Fig. 2(a)). Tin was welded on two points of the fiber bundle with spacing of 20 mm as contact electrodes. The measurements were carried out at room temperature after remained in 20 °C and 65% humidity for 24 h. The corresponding conductivity (σ) was calculated according to the following eqn (1):
 | (1) |
 | ||
| Fig. 2 (a) Electrical conductivity measurement setup consisting of a four-point probe and source meter; (b) a close-up image of the two-point method; (c) schematic illustration of measuring the electrical conductivity of carbon fibers bundle; and (d) transverse conductivity of Ag NPs-loaded carbon fibers. | ||
2.4. Mechanical property tests of composite
To further investigate the interfacial property between the fiber and the matrix, we carried out a microbond test to determine the interfacial shear strength (IFSS) of the Ag NPs-loaded carbon fiber/epoxy resin composite. The specimens of the microbond test were prepared in metal frames with dimension of 26 × 58 mm. The free fiber length was approximately 30 mm. Some epoxy resin droplets were placed against an Ag NPs-loaded carbon fiber monofilament and cured using the following curing process: 90 °C for 2 h, 120 °C for 2 h and 150 °C for 4 h. The microbond tests were performed on an interfacial strength testing machine (Tohei Sanyon Co., Ltd, Japan) at a crosshead displacement rate of 0.06 mm min−1 (illustration shown in Fig. 3(a) and (b)). The IFSS was calculated according to eqn (2).
 | (2) |
 | ||
| Fig. 3 (a) and (b) Schematic diagram of single fiber pull-out testing setup, not to scale. Variations in interfacial shear strength with (c) deposition time, (d) average Ag NPs size and (e) morphology of Ag NPs. (f) SEM of sphere Ag NPs-loaded carbon fiber and (g) dendritic Ag NPs-loaded carbon fiber. | ||
All the measurements were carried out at room temperature.
3. Results and discussion
3.1. Surface topography of carbon fibers
Fig. 4(a) shows the SEM image of the carbon fiber treated with electro-chemical deposition. Obviously, Ag NPs were planted on the surface of the carbon fiber and well distributed. The insert (Fig. 4(b)) shows the TEM image of the corresponding cross section of the Ag NPs-loaded carbon fiber. From the TEM image, we found the thickness of the silver coating was about 50–100 nm. Carbon fibers treated with the electro-chemical deposition for different deposition times were also studied, as shown in Fig. 5. At the beginning, the surface of untreated carbon fibers seemed to be very neat and smooth (Fig. 5(a)). Then, silver particles were plated on the carbon fibers and their size increased with reaction time at the expense of the smaller particles according to Ostwald ripening.29 The particle size grew to around hundreds of nanometers after a deposition time of 10 min. To clearly characterize the distribution and density of Ag NPs plated on the carbon fibers, an EDX mapping test was applied and results are presented as inserts in Fig. 5(b–d). It is clear that the density of Ag NPs became higher with the time growth. Meanwhile, the distribution of the Ag NPs stayed homogeneous. | ||
| Fig. 4 SEM (insert TEM) of the prepared Ag NPs-loaded carbon. | ||
 | ||
| Fig. 5 SEM images of carbon fibers: (a) untreated carbon fibers, (b) CF/Ag-3, (c) CF/Ag-4 and (d) CF/Ag-5; the corresponding EDX mapping images are inserted in (c and d). | ||
In the present study, spherical and dendritic silver coatings were prepared using the polyol process to control the morphology of the Ag NPs. Briefly, Ag NPs were produced by reducing silver nitrate with ethylene glycol in the presence of PVP via a polyol process.30 Without PVP, Ag NPs deposited on the carbon fiber aggregated, resulting in uneven morphology (as shown in Fig. 6(a)). A dendritic silver coating was obtained when the molar ratio of PVP to AgNO3 was 1.5 (Fig. 6(b)). When the molar ratio was increased from 1.5 to 3, spherical Ag NPs were the major product (Fig. 6(c)). It was speculated that the kinetics of adsorption and desorption of PVP on different crystallographic planes of the nanoparticles were different, which led to different morphologic structures.31 The AFM results of Ag NPs-loaded carbon fibers prepared with different molar ratios of PVP to AgNO3 (CF/Ag-6, CF/Ag-7 and CF/Ag-4) are shown in Fig. 6(d)–(f). Remarkable differences in the surface morphology and surface roughness were observed. The morphology of silver particles plated on carbon fibers was in accordance with the SEM results. In terms of the surface roughness, it was clear that Ra and Rmax of CF/Ag-6 were the highest among the three samples, because the silver particles aggregated easily when there was no PVP. Compared to CF/Ag-6, no obvious aggregation was found on the silver coatings of CF/Ag-7 (dendritic Ag NPs) and CF/Ag-4 (sphere Ag NPs). Especially, the CF/Ag-4 had the smallest Rmax and Ra (26.65 nM and 25.66 nM), meaning that the silver particles were distributed uniformly. These uniform Ag NPs could serve as a supplementary reinforcement to the interface and further reduced the stress concentration, as well as greatly enhanced the mechanical interlocking between the carbon fiber and the polymer matrix.
 | ||
| Fig. 6 SEM images of carbon fiber (a) CF/Ag-6, (b) CF/Ag-7 and (c) CF/Ag-4. AFM images of carbon fiber (d) CF/Ag-6, (e) CF/Ag-7 and (f) CF/Ag-4. | ||
3.2. XRD analysis
The typical XRD patterns of CF/Ag-7, CF/Ag-4 and virgin carbon fiber are shown in Fig. 7. Compared with the virgin carbon fiber, the XRD pattern of the two Ag NPs-loaded carbon fibers displays four peaks at about 38.1°, 44.2°, 64.4° and 77.4°, which are attributed to the crystal planes (111), (200), (220), and (311) diffraction peaks of face-centered cubic (FCC)-Ag, respectively. This revealed that FCC crystalline structure of Ag NPs were formed on the carbon fiber according to silver-3C (JCPDS card no. 04-0783). Meanwhile, the diffraction peaks of the crystal planes (111) and (200) indicated that there were no impurities, such as Ag2O and AgNO3, in the deposited Ag NPs. The peak intensity ratios of (111) and (200) were 4.1 and 2.2 for dendritic Ag NPs and spherical Ag NPs, respectively. For the dendritic Ag NPs, this ratio was higher than the standard file (JCPDS), which indicates that the dendritic Ag NPs were abundant in (111) facets, probably because of their external morphology. Besides, the peaks at 25.6° belong to unchanged amorphous carbon, indicating that the electro-chemical deposition treatment did not affect the carbon crystallographic structure of the carbon fibers. | ||
| Fig. 7 XRD patterns of carbon fiber, Ag/CF-7, Ag/CF-4 and silver-3C (JCPDS card no. 04-0783) are given as guides to the eye at the bottom. | ||
3.3. XPS analysis
XPS was performed to determine the chemical composition of the carbon fiber surface. The XPS spectra and the quantitative analysis are shown in Fig. 8 and Table 2. It is well known that the bonding energies for carbon, oxygen and silver are centered at around 284.6, 532.2, and 368.1 eV, respectively. From the experimental XPS results, it was found that the surface composition of the Ag NPs-loaded carbon fibers changed substantially. Fig. 8(a) shows the full spectra of carbon fibers before and after silver deposition. The silver element was found on the Ag NPs-loaded carbon fibers and the content was about 11.21%, which indicated the successful deposition of Ag on the carbon fibers. The detailed Ag 3d spectrum of the Ag NPs-loaded carbon fiber is shown in Fig. 8(b), from which we can see that Ag 3d 5/2 and Ag 3d 3/2 peaks are located at 368.32 eV and 374.27 eV, respectively, whereas the spin–orbit splitting of the 3d doublet is 5.95 eV. This bonding energy proved that silver on the carbon fibers surface is metallic silver (Ag0) in nature. The existence of Ag NPs on the carbon fibers was expected to improve the surface energy and wettability of the Ag NPs-loaded carbon fibers. | ||
| Fig. 8 XPS spectra of untreated and Ag NPs-loaded carbon fibers: (a) full spectra, (b) high-resolution spectra of Ag 3d regions of Ag NPs-loaded carbon fibers; XPS C 1s spectra of carbon fibers (c) untreated and (d) Ag NPs loaded. | ||
| Samples | C (%) | O (%) | Ag (%) |
|---|---|---|---|
| Pristine carbon fibers | 87.37 | 12.63 | 0 |
| AgNPs-loaded carbon fiber (sample 6) | 72.64 | 16.15 | 11.21 |
In order to calculate the ratio of functional groups on the fiber surfaces, the C 1s peaks of untreated and Ag NPs-loaded carbon fibers were fitted and the fitting results are shown in Fig. 8(c) and (d). By deconvolution of the C 1s region of the X-ray photoelectron spectra (XPS), five peaks representing graphitic carbon (peak I, 284.6 eV), carbon in phenolic, alcoholic, ether groups (peak II, 286.1–286.3 eV), carbonyl or quinone groups (peak III, 287.3–287.6 eV), carboxyl or ester groups (peak IV, 288.4–288.9 eV) and carbonate groups (peak V, 290.4–290.8 eV) were determined.32 Owing to the large functional groups on the virgin fiber surfaces, like alcoholic, carboxyl or ester groups, carbon fibers have high electronegativity in contrast to the π-conjugated bond. Meanwhile, these functional groups have the highest occupied molecular orbital (HOMO) and they prefer to transfer from the end-capping electron-donor groups to the electron-acceptor group (Ag+).33 In the case of Ag+ ions, empty electronic orbit and lowest unoccupied molecular orbital (LUMO) made their own high electrophilicity index. Compared with the π-conjugated bond on the fiber surfaces, Ag+ ions preferred to be adsorbed and reduced by functional groups. The schematic diagram in Fig. 9 illustrates the formation process of Ag NPs on the carbon fiber surface. A small number of Ag+ ions were reduced by the functional groups following the reactions in eqn (3) and (4), while it is believed that most of the Ag+ ions were reduced to silver according to eqn (5). So, there was no obvious difference between the C 1s peaks of the carbon fiber before and after deposition except that the carboxyl group content increased a little bit, which is consistent with the results of the C 1s spectra. The Ag+ ions were first reduced to form seeds of Ag crystals. Then, more Ag+ ions were reduced to enlarge the seeds, leading to the formation of Ag NPs, and the interaction between the Ag NPs and supporting carbon fibers would directly lead to the tight adherence.
 | (3) |
 | (4) |
 | (5) |
 | ||
| Fig. 9 Schematic illustration for fabricating Ag NPs-loaded carbon fibers. | ||
Based on the above results, the Ag NPs-loaded carbon fibers material in this study was postulated to form through the contact of the AgNO3 precursor solution and the carbon fiber surface. The Ag+ ions interacted with the hydroxyl and or carboxylic groups on the carbon fiber surface, and then the Ag+ was reduced to metallic Ag.
From the SEM, TEM, XRD and XPS results, we can reach the conclusion that the crystalline Ag NPs were successfully synthesized on the carbon fiber surface via a simple electro-chemical deposition process.
3.4. Single fiber tensile testing
To inspect the enhancing effect of the Ag modification, we investigated the tensile properties of the spherical Ag NPs-loaded fibers modified under different conditions (deposition time and Ag+ ion concentration), which affected the in-plane properties of the resulting carbon fiber/resin composites. The results of tensile strength of Ag NPs-loaded carbon fibers prepared with different deposition times are shown in Fig. 10(a). The tensile strength went from 4.06 GPa to 4.25 GPa as the deposition time went from 2 min to 10 min, indicating that the tensile strength increased with the deposition time. It is clear that the tensile strength of all Ag NPs-loaded carbon fibers was greater than that of the untreated carbon fiber (3.34 GPa). It was speculated that the Ag NPs filled the surface cracks on the surface of carbon fibers and effectively increased the crack tip radius, which reduced the stress concentration at the defects. Particularly, the tensile strength of modified carbon fiber treated for 5 min (4.21 GPa) was 27.2% higher than that of the untreated carbon fiber. The tensile strength of the fibers treated for 10 min was slightly higher than that of the fibers treated for 5 min, indicating that the surface defects of carbon fibers were almost fully covered by silver coating when the carbon fibers were treated for 10 min and the tensile strength of carbon fiber did not increase much with the longer deposition time. | ||
| Fig. 10 Variations in single fiber tensile strength with (a) deposition time and (b) morphology of Ag NPs. | ||
Fig. 10(b) shows the tensile strength of Ag NP-loaded carbon fibers with different morphologies. At the low Ag+ ions concentration, the tensile strength of spherical Ag NPs-loaded carbon fibers was bigger than that of dendritic ones. This implied that the distribution of spherical Ag NPs-loaded fibers was more even than that of dendritic ones. However, when the concentration of Ag+ ions was 30 mmol L−1, the tensile strengths of spherical and dendritic Ag NPs-loaded carbon fibers showed no obvious difference. The results revealed that the morphology of Ag NPs didn't affect the tensile strength at high concentration, because there were excessive Ag NPs on the carbon fiber no matter what the morphology was.
3.5. Interfacial property testing
To evaluate the influence of silver coating on the interfacial property of carbon fiber/resin composites, the interfacial shear strength (IFSS) was tested and is demonstrated in Fig. 3(c)–(g). The IFSS could truly reflect the interfacial bonding between the carbon fiber and resin (epoxy in this work) as it was not affected by external factors. The IFSS of pristine carbon fiber, CF/Ag-3, CF/Ag-4 and CF/Ag-5 with epoxy were 59.69, 75.77, 93.84 and 88.64 MPa, respectively (Fig. 3(c)). It was clear that the presence of Ag NPs influenced the IFSS of the carbon fiber and epoxy greatly. The density of Ag NPs increased as time went on, but the IFSS was not enhanced with the density of Ag NPs. For CF/Ag-3, Ag NPs were not compact enough, resulting in relatively weak mechanical interlocking. The mechanical interlocking increased with the content of Ag at the first stage and the maximum value existed in the interface between CF/Ag-4 and epoxy. As the content of Ag continued to increase, silver NPs aggregated on carbon fibers like CF/Ag-5, leading to deterioration of interfacial properties, which might be caused by the stress concentration and further formed defect areas.In addition, the size of Ag NPs also had an effect on the IFSS of carbon fiber/resin composites. Nano Measure 1.2 software was used to measure the diameters of 200 randomly selected spherical Ag NPs, then the diameters were analyzed using Gauss formulas to export the average diameter of the Ag NPs. Fig. 3(d) displays the relations between the IFSS of modified CF/epoxy composites and the diameters of spherical Ag NPs on carbon fibers. Four samples of Ag NPs modified CF with diameters of 18.20, 64.03, 98.50 and 152.79 nm were chosen and used to do the IFSS test. The IFSS of the four composites were found to be 75.77, 93.84, 87.32 and 82.06 MPa, respectively IFSS was enhanced with the enlargement of the diameter of Ag NPs. The increased diameter improved the roughness of the fiber surface. The surface roughness could effectively enhance the mechanical interlocking between the fiber and matrix. But when the diameters of Ag NPs were 98.50 and 152.79 nm, IFSS declined. That could be explained by the aggregation of the Ag NPs, which resulted in stress concentration.
Interestingly, we noted that spherical and dendritic silver coatings on the carbon fibers caused different IFSS improvement, as shown in Fig. 3(e). When the concentration of Ag+ ions was 6 mmol L−1, IFSS of the spherical Ag NPs-loaded carbon fiber/epoxy composite was 93.84 MPa, which was slightly higher than that of the dendritic Ag NPs-loaded carbon fiber/epoxy composite (91.15 MPa). This was because silver particles of the dendritic coating aggregated, which would cause stress concentration, and accordingly weakened the IFSS. However, when the concentration of Ag+ ions was 30 mmol L−1, the IFSS of the spherical and dendritic Ag NPs-loaded carbon fibers/epoxy composites both decreased and the values were close. This might be explained by the Ag NPs aggregation at high concentration of Ag+. From the data in Fig. 3(e), it could be seen that the IFSS was highly dependent on the quantity and distribution of Ag NPs in the interfacial region. The silver coating could change not only the surface morphology of the carbon fibers, but also the surface roughness, which could effectively enhance the mechanical interlocking between the fiber and matrix. He et al.34 reported that good dispersion and homogeneous distribution of Ag NPs were highly effective in increasing roughness, which resulted in improvement of the strength and toughness of the polymer composites.
In addition, the pictures of epoxy droplets before and after interfacial de-bonding were examined, as shown in Fig. 11. Fig. 11(a) shows the epoxy droplet image before de-bonding. Notably, the epoxy droplet was completely intact. After interfacial de-bonding, the epoxy droplet broke away from its home position. In the case of untreated carbon fiber (Fig. 11(b)), the de-bonded fiber surface was extremely smooth, on which there were only several epoxy fragments, indicating that the material failure mode was adhesive failure. With Ag NPs-loaded carbon fiber (Fig. 11(c) dendritic silver coating and Fig. 11(d) spherical silver coating, indicated by the green arrows), some of Ag NPs were bequeathed on the fiber surface. This also demonstrated that the adhesive force between Ag NPs and carbon fiber was so strong that Ag NPs did not move with epoxy droplets. Moreover, there were many scratches on the fiber surfaces (marked by the green rectangles in Fig. 11(c) and (d)), indicating that Ag NPs filled in the crack and microhole of the carbon fiber scratched the carbon fiber when they were moved with the epoxy. For the excellent distribution of silver particles on the fiber surface, the interface was able to transfer load and consume impact energy better. The improvement of the interfacial strength could be attributed to the enhancement of the mechanical interlocking and the increased fiber surface area caused by Ag NPs on the fiber surface. These Ag NPs loaded on the fiber surface stuck into the epoxy matrix and worked as an anchor to locally stiffen at the interface region. This made clear that the anchoring strength of the Ag-loaded carbon fiber/epoxy matrix was much stronger than that of the untreated carbon fiber/epoxy matrix.
 | ||
| Fig. 11 Surface morphology of (a) an epoxy droplet before debonding, (b) untreated, (c) dendritic Ag loaded and (d) sphere Ag loaded carbon fiber composites after de-bonding. | ||
3.6. Electrical conductivity
Fig. 2(d) displays the average electrical conductivity of Ag NPs-loaded carbon fibers with different process parameters. It is clear that the electrical conductivity of every Ag NPs-loaded carbon fiber was better than that of the untreated carbon fiber (29.3 μS cm−1). The presence of highly conductive silver particles formed new conducting paths on the carbon fibers to achieve improvement of the conductivity. The maximum electrical conductivity of the Ag NPs-loaded carbon fibers was 79.9 μS cm−1, which was about a 2-fold enhancement as compared to that of the virgin carbon fibers. The electrical property improvement of 30% was also reported in the case of carbon fibers covered with carbon nanotubes by electrophoresis.35The average electric conductivities, σ, of Ag/CF-4 and Ag/CF-5 were 59.9 and 72.2 μS m−1. We speculated that the difference in the electrical conductivity was caused by the thicknesses difference of the composite specimens. The difference between Ag/CF-4 and Ag/CF-7 may also originate from the different morphologies of the deposited silver particles. As opposed to the dendritic structure, the spherical silver particles were predominantly deposited dispersedly and uniformly, thus exerting a higher electrical conductivity for the formation of an interconnected percolating network on the carbon fibers.
This study demonstrated that the deposition of a small amount of silver particles was sufficient to induce a significant improvement in the electrical conductivity of the carbon fibers as a result of the reinforcement of the electron transport channels, and further enhancement was anticipated with an increase in silver loading. This improvement was particularly important and desirable for the conductive composites, which potentially provided a sufficient lightning current pathway to protect the composite airframes.36,37
3.7. Improvement mechanism
The strong anchoring strength between the Ag NPs and carbon was another reason for the improvement of interfacial properties and single fiber tensile strength. Theoretical calculation was carried out with periodic boundary conditions using Materials Studio 6.0 (from Accelrys Inc.).38–40 This can evaluate the anchoring strength between fiber and Ag NPs. In the calculation system, the carbon (C) molecule consisted of 68 carbon atoms with some surface oxygenic functional groups, while the Ag molecule consisted of 50 Ag atoms with the minimum cell unit cleaved along the (111) facet (the results are shown in Fig. 12). All the calculations were carried out using the Forcite module with the COMPASS force field. | ||
| Fig. 12 Theoretical calculation result of Ag NPs-loaded carbon fiber with different perspectives: (a) top view and (b) side view. | ||
The interaction energy between Ag and C (ELFP-GO, kJ mol−1) was calculated using the following equation:
| Einter = Ec + EAg − Etotal |
According to this formula, the interaction energy was 3.02 × 105 kJ mol−1 (222.29 MPa), which was much bigger than the IFSS of the carbon fiber/epoxy composite. Thus, most of the Ag NPs stayed on the fiber surface and worked as an anchor to improve the mechanical interaction. The Ag NPs interphase worked as a shielding layer, which could relieve the stress concentration, thus preventing the crack tips from direct contact with the fiber surface and making the crack path deviate away from the fiber surface to the interphase region.
At the same time, the massive improvement in fiber tensile strength was attributed to Ag NPs strong adhesive force and “bridges” built by the silver coating on the defect tips on the fiber surface, which in turn delay the crack opening. A schematic illustration is shown in Fig. 9, which gives insight into the healing effect of the crack and microhole on the carbon fiber surface. Through optimizing the interphase structure, the interfacial strength and tensile strength could be simultaneously improved.
4. Conclusions
In summary, we have developed a high-efficiency and green electro-chemical deposition approach to synthesize various structures of Ag NPs on carbon fibers by controlling the reactant ratios, deposition time and concentrations. The presence of PVP as the surfactant was proved to be important for the preparation of homogeneous Ag NPs. The Ag NPs modification simultaneously improved the single fiber tensile strength of the carbon fiber and the IFSS of its composite to a maximum of 0.91 GPa and 34.15 MPa, respectively. Furthermore, it is demonstrated that the mechanical property of the spherical silver coating was better than that of the dendritic one owing to the perfect dispersity and the uniform particle size. We also found that Ag NPs-loaded carbon fibers have excellent conductivity, benefiting from the high electrical conductivity of metallic silver. Owing to the possibility of scalable synthesis and prominent properties, the Ag NPs-loaded carbon fibers could offer attractive opportunities in both fundamental study and potential industrial applications, for instance, catalysis, adsorption, separation, conduction and composite materials.Acknowledgements
The authors gratefully acknowledge the National High Technology Research and Development Program of China (863 Program, grant number 2012AA03A212), International S&T Cooperation Program of China (2013DFR40700) and National Natural Science Foundation of China (51203034).References
- F. Zhao, Y. D. Huang, L. Liu, Y. P. Bai and L. W. Xu, Carbon, 2011, 49, 2624 CrossRef CAS.
- L. C. Ma, L. H. Meng, D. P. Fan, J. M. He, J. L. Yu and Y. D. Huang, Appl. Surf. Sci., 2014, 61, 296 Search PubMed.
- Y. W. Wang, L. H. Meng, L. Q. Fan, G. S. Wu, L. C. Ma and Y. H. Huang, RSC Adv., 2015, 5, 44492–44498 RSC.
- H. L. Cao, Y. D. Huang, Z. Q. Zhang and J. T. Sun, Compos. Sci. Technol., 2005, 65, 1655 CrossRef CAS.
- C. U. Jr Pittman, G. R. He, B. Wu and S. D. Gardne, Carbon, 1997, 35, 317 CrossRef.
- M. A. Montes-Morán, F. W. J. van Hattum, J. P. Nunes, A. MartínezAlonso, J. M. D. Tascón and C. A. Bernardo, Carbon, 2005, 43, 1795 CrossRef.
- Z. W. Xu, Y. D. Huang, C. H. Zhang, L. Liu, Y. H. Zhang and L. Wang, Compos. Sci. Technol., 2007, 67, 3261 CrossRef CAS.
- I. Lópeza, A. Vázqueza, G. H. Hernández-Padrónb and I. Gómeza, Appl. Surf. Sci., 2013, 280, 715 CrossRef.
- S. Q Zhu, T. Zhang, X. L. Guo, Q. L. Wang, X. F. Liu and X. Y. Zhang, Nanoscale Res. Lett., 2012, 7, 613 CrossRef PubMed.
- W. Fan, Y. Wang, J. Chen, Y. Yuan, A. Li, Q. Wang and C. Wang, RSC Adv., 2015, 5, 75735–75745 RSC.
- R. Sharma and K. K. Kar, RSC Adv., 2015, 5, 66518–66527 RSC.
- M. Montazer and V. Allahyarzadeh, Ind. Eng. Chem. Res., 2013, 52, 8436 CrossRef CAS.
- L. Chen, Z. Hu, L. Xing, J. Wu, N. Zheng, L. Liu, J. He and Y. D. Huang, Fibers Polym., 2014, 15, 1160–1167 CrossRef CAS.
- F. Zhao and Y. D. Huang, J. Mater. Chem., 2011, 21, 3695–3703 RSC.
- Y. Soo-Jin Park, S. Jang and R. Kyong-Yop, J. Colloid Interface Sci., 2002, 245, 383–390 CrossRef PubMed.
- L. Y. Yua, Q. Zhang, Q. Xu, D. Q. Jin, G. D. i. Jin, K. X. Li and X. Y. Hu, Talanta, 2015, 143, 245–253 CrossRef PubMed.
- J. Du, R. R. Yue, F. F. Ren and Z. Q. Yao, Gold Bull., 2013, 46, 137–144 CrossRef.
- C. Tang, W. Sun and W. Yan, RSC Adv., 2014, 4, 523–530 RSC.
- H. Chien-Te, C. Pan and W. Y. Chen, J. Power Sources, 2011, 196, 6055–6061 CrossRef.
- S. Kwon, R. Ma, U. Kim, H. R. Choi and S. Baik, Carbon, 2014, 68, 118–124 CrossRef CAS.
- Y. G. Sun and Y. N. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- F. Zhao and Y. D. Huang, Carbon, 2011, 49, 2624–2632 CrossRef CAS.
- W. Song, A. J. Gu, G. Z. Liang and L. Yuan, Appl. Surf. Sci., 2011, 257, 4069–4074 CrossRef CAS.
- W. Z. Qin and F. Vautard, Composites, Part A, 2015, 69, 335–341 CAS.
- Z. S. Wu, S. F. Pei, W. C. Ren, D. M. Tang, L. B. Gao, B. L. Liu, F. Li, C. Liu and H. M. Cheng, Adv. Mater., 2009, 21, 1756–1760 CrossRef CAS.
- P. Sarkar, S. Datta and S. Patrick, Composites, Part A, 1997, 28, 49–56 Search PubMed.
- V. Siva Kumar, B. M. Nagaraja, V. Shashikala, A. H. Padmasri, S. Shakuntala Madhavendra, B. David Raju and K. S. Rama Rao, J. Mol. Catal. A: Chem., 2004, 223, 313–319 CrossRef.
- L. Guadagno, U. Vietri, M. Raimondo, L. Vertuccio, G. Barra, B. De Vivo, P. Lamberti, G. Spinelli, V. Tucci, F. De Nicola, R. Volponi and S. Russo, Composites, Part B, 2015, 80, 7–14 CrossRef CAS.
- Y. G. Sun, B. Mayers, T. Herricks and Y. N. Xia, Nano Lett., 2003, 3, 955–960 CrossRef CAS.
- T. C. Deivara, N. L. Lala and J. Y. Lee, J. Colloid Interface Sci., 2005, 289, 402–409 CrossRef PubMed.
- Y. G. Sun and Y. N. Xia, Adv. Mater., 2002, 14, 833 CrossRef CAS.
- S. Y. Huang, G. P. Wu, C. M. Chen, Y. Yang, S. C Zhang and C. X. Lu, Electrophoretic deposition and thermal annealing of a graphene oxide thin film on carbon fiber surfaces, Carbon, 2013, 52, 613–616 CrossRef CAS.
- C. Yohannan Panicker, H. Tresa Varghese, B. Narayanac, K. Divya, B. K. Sarojini, J. Ahmad War, C. Van Alsenoy and H. K. Fung, J. Mol. Catal. A: Chem., 2015, 148, 29–42 Search PubMed.
- J. M. He and Y. D. Huang, Polym. Polym. Compos., 2006, 14, 123–133 CAS.
- E. Bekyarova, E. T. Thostenson, A. Yu, H. Kim, J. Gao, J. Tang, H. T. Hahn, T.-W. Chou and M. E. Itkis, Langmuir, 2007, 23, 3970–3974 CrossRef CAS PubMed.
- M. C Guo, X. S. Yi, G. Liu and L. P. Liu, Compos. Sci. Technol., 2014, 97, 27–33 CrossRef.
- D. D. L. Chung, Carbon, 2012, 50, 3342–3353 CrossRef CAS.
- W. Zhang, F. K. Yang, Z. Pan, J. Zhang and B. Zhao, Macromol. Rapid Commun., 2014, 35, 350–354 CrossRef CAS PubMed.
- B. Wang, A. M. Liu, W. Al Abdulla, D. L. Wang and X. S. Zhao, Nanoscale, 2015, 7, 8819–8828 RSC.
- Z. S. Feng, C. Zhang, J. J. Chen, Y. Wang, X. Jin, R. Zhang and J. Hu, RSC Adv., 2013, 3, 4408 RSC.
| This journal is © The Royal Society of Chemistry 2016 |