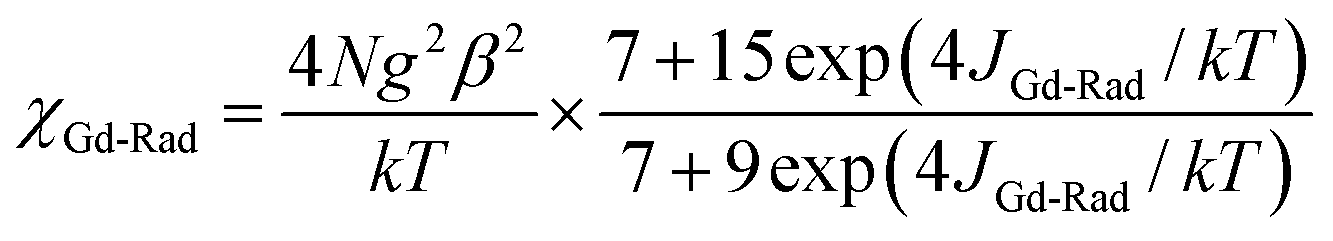DOI:
10.1039/C5RA21991D
(Paper)
RSC Adv., 2016,
6, 3058-3067
Modulating spin dynamics of LnIII-radical complexes by using different coligands†
Received
27th October 2015
, Accepted 15th December 2015
First published on 18th December 2015
Abstract
The combination of LnIII ions (GdIII, TbIII or DyIII) with a triazole nitronyl nitroxide radical results in six novel 2p–4f compounds, namely, [Ln2(hfac)6(MeTrzNIT)(H2O)2]·1/2CH2Cl2 (Ln = Gd(1), Tb(2), Dy(3); hfac = hexafluoroacetylacetone; MeTrzNIT = 2-[3-(5-methyl-l,2,4-triazolyl)]-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide and [Ln(Phtfac)3(MeTrzNIT)]2·C7H14·3H2O (Ln = Gd(4), Tb(5), Dy(6); Phtfac = 4,4,4-trifluoro-1-phenylbutane-1,3-dione). Single crystal X-ray diffraction studies revealed that compounds 1–3 are binuclear isostructural complexes, in which one MeTrzNIT molecule acts as a double-bridging ligand coordinated to two LnIII ions through its two NO groups and two nitrogen atoms of the triazole ring. In 1–3, the coordination number around the lanthanide ion is nine, and the polyhedron is a 4,4,4-tricapped trigonal prism (D3h). While the larger steric hindrance of the Ph- group than CF3- in the Phtfac ligand induces complexes 4–6 to be mononuclear bi-spin compounds, in which central LnIII ions are coordinated by three Phtfac and one bidentate MeTrzNIT radical. The coordination number around the lanthanide ion in 4–6 is eight, and the polyhedron is in a square antiprism geometry (D4d). Compounds 3 and 5 were found to exhibit slow relaxation of the magnetization, suggesting single-molecule magnet (SMM) behavior, while no ac signal is noticed for compounds 2 and 6. The different magnetic relaxation behaviours between 2 and 5, or between 3 and 6, are due to the different crystal structures around the LnIII ions and the magnetic interaction. It is demonstrated that the β-diketonate coligand may play an important role in determining the spin dynamics for the lanthanide-radical system.
Introduction
Designing and synthesizing of low-dimensional assemblies based on anisotropic metal ions that show magnetization relaxation have attracted much attention.1,2 Such materials named as single-molecular magnets (SMMs) and single chain magnets (SCMs), have potential applications in high-density data storage, quantum information processing systems, and spintronic devices.3–5 The general character of SMMs is that the magnetic bistability arises from the blocking anisotropy without long-range ordering. One of the challenging problems in this field is to increase the blocking temperature at which superparamagnetic behavior can occur, which depends on the anisotropy barrier from a combination of the appropriate spin in the ground state and uniaxial magnetic anisotropy.6 Lanthanides ions, especially heavy lanthanide ions, have large number of unpaired f-electrons and large intrinsic magnetic anisotropy, and have become good candidates for the construction of SCMs and SMMs.7–10 But, the naturally accompanying quantum tunneling from the hyperfine couplings and dipolar spin–spin interactions of lanthanide ions always lowers the effective relaxation energy barrier and induces the loss of remnant magnetization.11 Recent studies show that strong coupling through a radical bridge (with a record blocking temperature) and strong axiality or Ising exchange interaction can suppress quantum tunneling to provide strategies for enhancing the SMM properties.12 Nitronyl nitroxide radicals (NITs) as spin carriers are fascinating building blocks and bridges not only for their stabilization under ambient condition but also for the π systems to transfer the effective magnetic interactions. The use of organic radicals has been proved to be an attractive route to obtain magnetically coupled 4f-organic radical heterospin systems.13,14
For SMMs containing lanthanide ion, magnetic relaxation is very sensitive to the symmetry of the ligand field of the rare earth ion and the spin dynamic can be modified by the careful adjustment of the ligand field around the metal center. In order to explore how the symmetry of the local crystal field around the lanthanide center affect the spin dynamics of the complex, we decided to use two β-diketonate coligands hexafluoroacetylacetonate (hfac) and 4,4,4-trifluoro-1-phenylbutane-1,3-dione (Phtfac) to construct radical-lanthanide SMMs. The introduction of a Ph-group in the β-diketonate ligand may modify the ligand field of the metal ion, thus adjusting the magnetic relaxation of the molecule. Herein we synthesized six lanthanide compounds based on a triazole nitronyl nitroxide radical (MeTrzNIT = 2-[3-(5-methyl-l,2,4-triazolyl)]-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide) (Scheme 1) and coligand hfac or Phtfac, namely, [Ln2(hfac)6(MeTrzNIT)(H2O)2]·1/2CH2Cl2 (Ln = Gd(1), Tb(2), Dy(3)) and [Ln(Phtfac)3(MeTrzNIT)]2·C7H14·3H2O (Ln = Gd(4), Tb(5), Dy(6)). Magnetic studies showed that complexes 3 and 5 exhibit frequency-dependent ac susceptibility at low temperature, which suggest SMMs behavior. The comparison of the magnetic properties of 2 and 5 or 3 and 6 highlights that the β-diketonate ligands can play an important role in modulating the magnetic relaxation.
 |
| | Scheme 1 Structure of the triazole nitronyl nitroxide radical ligand. | |
Experimental details
Materials and physical measurements
All of the reagents used in the syntheses were of analytical grade, except that the solvents used were dried (heptane over sodium, CH2Cl2 over CaH2 and CHCl3 over P2O5) and distilled prior to use. The starting materials Ln(hfac)3·2H2O and Ln(Phtfac)3·2H2O were synthesized according to methods in the literature.15 MeTrzNIT = 2-[3-(5-methyl-l,2,4-triazolyl)]-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide was prepared based on the literature method.16 Elemental analyses for carbon, hydrogen, and nitrogen were performed on a Perkin-Elmer 240 elemental analyzer. Infrared spectra were recorded from KBr pellets in the 4000–400 cm−1 region on a Bruker TENOR 27 spectrometer. Powder X-ray diffraction measurements were recorded on a D/Max-2500 X-ray diffractometer using Cu-Kα radiation. Direct-current (dc) magnetic susceptibilities of crystalline samples were measured on an MPMS-7 SQUID magnetometer in the temperature range of 2–300 K with 1000 Oe applied magnetic field. Alternating-current (ac) susceptibilities were performed on the same magnetometer under zero static field with an oscillating of 3.5 Oe at frequencies up to 1500 Hz. The data were corrected for the diamagnetism of the samples using Pascal constants.
Preparation of complexes of 1–6
Complexes 1–3 were obtained by dissolving Ln(hfac)3·2H2O (0.1 mmol) (Ln = Gd (1), Tb (2), Dy (3)) in boiling n-heptane (20 mL). After stirring for 2 h, the solution was cooled to 60 °C, to which MeTrzNIT (0.05 mmol) in CH2Cl2 (5 mL) was added. The mixture was refluxed for 30 min. Then the solution was cooled to room temperature, filtrated and the filtrate was stored in a refrigerator at 0–4 °C for three or four weeks to give blue-violet crystals, which were suitable for X-ray analysis.
Complexes 4–6 were obtained by dissolving Ln(Phtfac)3·2H2O (0.1 mmol) (Ln = Gd(4), Tb (5), Dy (6)) in boiling n-heptane (30 mL). After stirring for 2 h, the solution was cooled to 60 °C, to which MeTrzNIT (0.1 mmol) in CH2Cl2 (5 mL) was added. The mixture was refluxed for 30 min. Then the solution was cooled to room temperature, filtrated and the filtrate was stored in a refrigerator at 0–4 °C for four or five weeks to give blue-violet crystals.
[Gd2(hfac)6(MeTrzNIT)(H2O)2]·1/2CH2Cl2 (1). Yield 0.068 g, 72%. C40.5H27ClF36Gd2N5O16 (1873.62): calcd C 25.96, H 1.45, N 3.74; found: C 25.54, H 1.32, N 3.55%. IR (KBr pellet): 2354 (w), 1662 (w), 1383 (m), 1150 (s), 1078 (vs), 960 (m), 863 (vs), 544 (vs) cm−1.
[Tb2(hfac)6(MeTrzNIT)(H2O)2]·1/2CH2Cl2 (2). Yield 0.064 g, 68%. C40.5H27Tb2ClF36N5O16 (1876.96): calcd for C 25.91, H 1.45, N 3.73; found: C 25.82, H 1.52, N 3.52%. IR (KBr pellet): 2352 (w), 1655 (w), 1386 (m), 1148 (vs), 1079 (vs), 973 (m), 848 (s), 538 (s) cm−1.
[Dy2(hfac)6(MeTrzNIT)(H2O)2]·1/2CH2Cl2 (3). Yield 0.059 g, 64%. C40.5H27ClDy2F36N5O16 (1884.13): calcd C 25.82, H 1.44, N 3.72; found: C 26.12, H 1.27, N 3.78%. IR (KBr pellet): 1663 (w), 1395 (w), 1153 (s), 1081 (vs), 952 (m), 859 (m), 553 (s) cm−1.
[Gd(Phtfac)3(MeTrzNIT)]2·C7H14·3H2O (4). Yield 0.071 g, 63%. C87H90F18Gd2N10O19 (2236.18): calcd C 46.73, H 4.06, N 6.27; found: C 46.52, H 3.68, N 6.04%. IR (KBr pellet): 3151 (vs), 1617 (s), 1578 (w), 1531 (w), 1459 (w), 1402 (s), 1290 (m), 1190 (s), 1139 (s), 776 (m), 704 (m), 631 (s) cm−1.
[Tb(Phtfac)3(MeTrzNIT)]2·C7H14·3H2O (5). Yield 0.078 g, 69%. C87H90F18Tb2N10O19 (2240.5): calcd C 46.64, H 4.05, N 6.25; found: C 46.41, H 3.81, N 6.13%. IR (KBr pellet): 3164 (vs), 1616 (s), 1572 (w), 1538 (w), 1469 (w), 1401 (s), 1291 (m), 1190 (s), 1140 (s), 762 (m), 703 (m), 626 (s) cm−1.
[Dy(Phtfac)3(MeTrzNIT)]2·C7H14·3H2O (6). Yield 0.072 g, 64%. C87H90F18Dy2N10O19 (2246.61): calcd C 46.50, H 4.04, N 6.24; found: C 46.16, H 3.75, N 6.31%. IR (KBr pellet): 3156 (vs), 1617 (s), 1578 (w), 1531 (w), 1467 (w), 1402 (s), 1291 (m), 1186 (s), 1139 (s), 762 (m), 698 (m), 631 (s) cm−1.
X-ray crystallography
The crystallographic data of compounds 1–4 were carried out with an Oxford Diffractometer SuperNova TM, which were equipped with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å). Lorentz polarization and absorption corrections were applied. Structures were solved by direct methods with the SHELXS-97 program and refined by full-matrix least-squares techniques against F2 with the SHELXTL-97 program package.17 Some restraints are applied, such as ISOR (anisotropic parameter), DFIX (restricting the distance between two atoms) to solve the disorder of the F atoms and CH2Cl2 in 2 and 3. Besides fluorine atoms, all other non-hydrogen atoms were refined anisotropically, and hydrogen atoms were located and refined isotropically. Although complex 4 successfully underwent X-ray analysis after many times' experiments, it couldn't give good crystallography data because the crystals are easy to efflorescence. The R1 and wR2 data of 4 are a little larger than the normal data. For complexes 5 and 6, the crystals are much easier to be weathered than 4 and they can't undergo X-ray analysis even at low temperature. Powder X-ray diffraction, elemental analysis, and infrared spectroscopy confirmed that complexes 5 and 6 are isomorphous to 4. Crystallographic data for the compounds 1–4 are listed in Table 1 and the powder X-ray diffraction data for all the six compounds are shown in the ESI section (Fig. S1 and S2†).
Table 1 Crystallographic data and structure refinement details for 1–4
| |
1 |
2 |
3 |
4 |
| Formula |
C40.5H27ClF36Gd2N5O16 |
C40.5H27Tb2ClF36N5O16 |
C40.5H27ClDy2F36N5O16 |
C87H90F18Gd2N10O19 |
| Mr |
1873.62 |
1876.96 |
1884.13 |
2236.15 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
C2/c |
C2/c |
C2/c |
C2/c |
| a (Å) |
25.752(4) |
25.7993(9) |
25.805(5) |
27.474(2) |
| b (Å) |
24.466(4) |
24.4562(9) |
24.464(5) |
21.7415(18) |
| c (Å) |
23.085(3) |
23.1575(9) |
23.177(5) |
33.465(3) |
| α (°) |
90 |
89.34(3) |
90 |
90 |
| β (°) |
120.758(4) |
120.85(2) |
120.81(3) |
102.2500(10) |
| γ (°) |
90 |
90 |
90 |
90 |
| V (Å3) |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 499(3) 499(3) |
12544.6(8) |
12567(4) |
19535(3) |
| Z |
8 |
8 |
8 |
8 |
| ρcalc (Mg m−3) |
1.991 |
1.988 |
1.947 |
1.414 |
| μ (mm−1) |
2.319 |
2.451 |
2.530 |
1.442 |
| F(000) |
7216 |
7232 |
7080 |
8272 |
| θ range (°) |
1.66–25.01 |
1.84–25.01 |
1.24–25.01 |
1.245–28.379 |
| GOF on F2 |
1.043 |
1.030 |
1.069 |
1.075 |
| R1, wR2 [I > 2σ(I)] |
0.0508, 0.1208 |
0.0383, 0.0766 |
0.0583, 0.1424 |
0.1082, 0.2385 |
| R1, wR2 (all data) |
0.0707, 0.1373 |
0.0518, 0.0834 |
0.0714, 0.1498 |
0.2163, 0.2911 |
Results and discussion
Crystal structure
Structure of 3. Single-crystal X-ray diffraction analyses reveal that complexes 1–3 are isomorphous and belong to monoclinic C2/c space group with Z = 8. In view of their structural similarity, only the structure of 3 will be described herein as a representative example. The structural diagrams of 1–2 are given in the ESI, Fig. S3 and S4.† As shown in Fig. 1, two Dy(hfac)3 units are connected by a radical ligand MeTrzNIT to give a binuclear core. The center ions are all surrounded with a slightly distorted 4,4,4-tricapped trigonal prism (D3h) DyO8N coordination sphere from three bischelate hfac anions and one bridging radical ligand. The Dy–O(N) (nitroxide) distances are 2.408(6) and 2.416(5) Å, respectively. The Dy–O(hfac) bond lengths are in the range of 2.337(6)–2.478(5) Å, the Dy–N3 and Dy–N5 distances are longer than the normal Dy–N bonds ascribed to the bridged character of N3 and N5 (Table S1†). Each MeTrzNIT acts as a double-bridging ligand and is coordinated to two different DyIII ions through the two oxygen atoms of NO groups and two nitrogen atoms of the triazole ring. The Dy⋯Dy separation distance in every binuclear unit is 7.277(5) Å and the shortest intermolecular Dy⋯Dy separation is 5.995(6) Å. Here the five membered imidazoline ring and the triazole ring show average twist angles of 16.6(2)°. The five-membered ring of the radical deviates significantly from planarity with C20 atom 0.045 Å above the average plane.
 |
| | Fig. 1 (a) Simplified view of the crystal structure of 3. Fluorine, hydrogen, and some carbon atoms are omitted for clarity. (b) Polyhedral representation of the Dy3+ cores. | |
Structure of 4. Compound 4 also crystallizes in space group C2/c with Z = 8. The asymmetric unit contains two crystallographically independent [Gd(Phtfac)3(MeTrzNIT)] moieties, and every [Gd(Phtfac)3(MeTrzNIT)] exhibits mononuclear structure with central metal ion in an LnO7N coordination sphere (Fig. 2). Powder X-ray diffraction, confirmed that complexes 5 and 6 are isomorphous to 4. As shown in Fig. 2a, each central GdIII ion is eight-coordinated with three bidentate β-diketonate coligands and one bidentate MeTrzNIT radical ligand. The Gd–O(Phtfac) distances range from 2.313(8) to 2.392(9) Å. The Gd–O(radical) and Gd–N(triazole) bond lengths of compound 4 are in the range of 2.365(8)–2.386(8) Å and 2.653(8)–2.697(8) Å, respectively. When applying the D4d symmetry to the GdO8 site, CSM method gives the Gd1 and Gd2 the minimal value of S = 0.434 or S = 0.621, respectively.
 |
| | Fig. 2 (a) Simplified view of the crystal structure of 5. Fluorine, hydrogen, and some carbon atoms are omitted for clarity. (b) D4d-symmetry polyhedral of gadolinium atoms. | |
Magnetic properties
Static magnetic properties for 1–3. Variable temperature magnetic susceptibilities for complexes 1–3 are studied and shown in Fig. 3. At room temperature, the χMT value is 16.53 cm3 K mol−1 for complex 1, which is close to the expected values of 16.29 cm3 K mol−1 for the isolated spins of two GdIII ions (8S7/2, g = 2) and one radical (S = 1/2, 0.375 cm3 K mol−1). On decreasing the temperature, the χMT value almost remains unchanged till 50 K. Then it begins to increase quickly as the temperature is lowered further and reaches a peak value of 19.14 cm3 K mol−1 at 2 K. The profile of the curve indicates that the GdIII ion and nitroxide radical interactions are ferromagnetic. Based on the above structural analysis, two main exchange pathways should be operative: (i) the magnetic interaction between Gd and the directly coordinated nitroxide group (J); (ii) the magnetic coupling between Gd(1) and Gd(2) through triazole ring (J′). Accordingly, the system was modeled as a tri-spin unit, and the magnetic analysis for GdIII-Rad unit was carried out by using the spin Hamiltonian Ĥ = −2J(ŜGd1ŜRad1 + ŜGd2ŜRad1) − 2J′ŜGd1ŜGd2. Eqn (S1) and (S2) (which is shown in ESI section†) is introduced to analyze the magnetic coupling strength, where J and J′ represent the magnetic coupling for Gd-radical and Gd1-Gd2, respectively. The weak exchange interaction being considered within the mean field approximation (zj′).18
 |
| | Fig. 3 Temperature dependence of χMT values for complexes 1–3. The solid line represents the theoretical values based on the corresponding equations. | |
The observed χMT data were well reproduced, giving the best fitting parameters of gGd = 1.99, J = 1.65 cm−1, and J′ = −0.1724 cm−1, zj′ = 0.0043 cm−1 with R = 1.38 × 10−3 (gR and g was fixed as 2). The positive values of J indicate the ferromagnetic interactions between GdIII ion and the radical, which is very common in Gd-radical complexes.19 The antiferromagnetic reaction J′ between the two GdIII ions through imidazoline and triazole rings is quite weak.20,21 The magnetization versus field measurements at 2 K is shown in Fig. 4 (left). A magnetization of 15.08 Nβ is reached at 50 kOe, in agreement with the 14.96 Nβ for the ferromagnetic arrangement for the spins.
 |
| | Fig. 4 Field dependence of the magnetization at 2 K for complex 1 (left) and 2 (right). | |
For 2 (Fig. 3), the value of χMT at 300 K is 24.41 cm3 K mol−1, which is a little higher than the expected value 23.84 cm3 K mol−1 for two uncoupled TbIII ions (7F6 and g = 3/2) and one organic radical (S = 1/2). At high temperature (130–300 K), the value of χMT almost remains unchanged. As the temperature is lowered from 130 K, χMT decreases to reach 19.43 cm3 K mol−1 at 2 K. The decrease of the χMT values in the high temperature regime is most probably governed by the depopulation of the TbIII Stark sublevels. The decrease of χMT at low temperature suggests the presence of intramolecular antiferromagnetic interaction. This magnetic behavior may be ascribed to the exchange interaction between the paramagnetic ions (Rad-Tb(III), Tb(III)–Tb(III) interaction through triazole ring) combined with the crystal field and spin–orbit effect. At present, it is not possible to quantify the different contributions,22 but the crystal field and spin–orbit effect may be dominant due to the completed 5s and 5p subshells shielding 4f electrons.23 The field dependences of magnetization (M) for complex 2 have been determined at 2 K in the range of 0–70 kOe (Fig. 4 (right)). The field-dependent magnetization values below 10 K show a rapid increase in the magnetization at low magnetic fields. The maximum magnetization is 11.8 Nβ at 2 K and 70 kOe, which does not reach the expected saturation values of 19 Nβ (18 Nβ for two TbIII ions for J = 6 and g = 3/2, plus 1 Nβ for an organic radical), most likely because of the crystal field effect on the TbIII ion.24
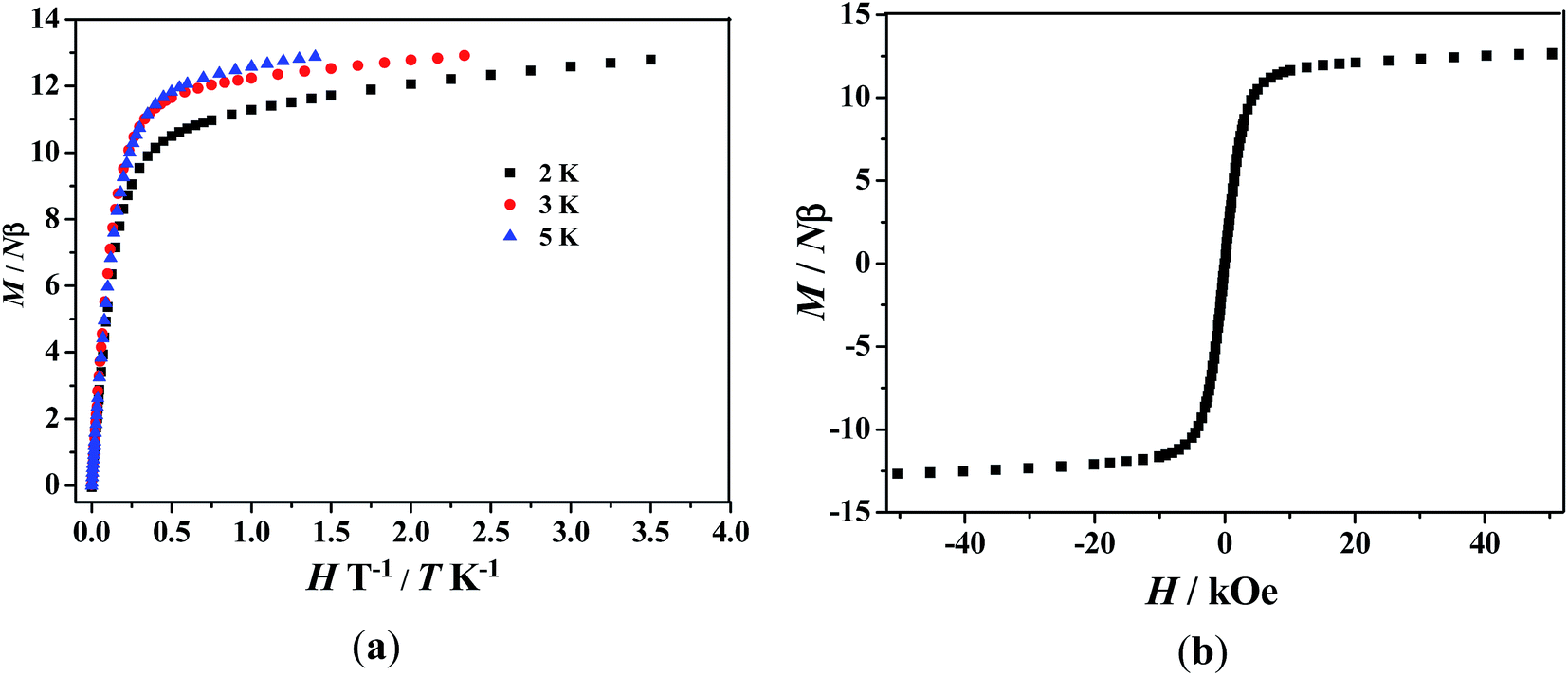
For 3, the value of χMT at room temperature is 28.38 cm3 K mol−1, which is slightly lower than the expected value of 28.70 cm3 K mol−1 for two isolated DyIII ions (6H15/2) and one organic radical (S = 1/2). As the temperature is lowered, χMT decreases slowly to reach a minimum of 25.97 cm3 K mol−1 at 20 K. Below 20 K, χMT increases dramatically to reach a peak value of 32.63 cm3 K mol−1 at 2 K. The decrease of χMT upon lowering of the temperature in the high-temperature range is most probably governed by depopulation of the LnIII Stark sublevels. The marked increase of χMT at low temperature indicates the presence of ferromagnetic interactions. The field dependences of magnetization (M) for complex 3 have been determined at 2–5 K in the range of 0–70 kOe (Fig. 5a). The field-dependent magnetization value below 5 K shows a rapid increase at low magnetic fields. At higher fields, M increases up to 12.79 Nβ at 2 K and 70 kOe, which does not reach the expected saturation values of 21 Nβ (10 Nβ for each DyIII ion for J = 15/2 and g = 4/3, plus 1 Nβ for the organic radical). The nonsuperposition on the M versus H/T curves at different temperatures indicates the presence of a magnetic anisotropy and/or low lying excited states in the system, which corresponds to the reported results.8b,9a,23,24,25,26 Fig. 5b shows the magnetization versus field curve at the temperature of 2 K, but no hysteresis loop was observed.
 |
| | Fig. 5 (a) Field dependence of the magnetization at different temperatures for complex 3. (b) Magnetization versus field measurement for complex 3 as a polycrystalline sample at 2 K. | |
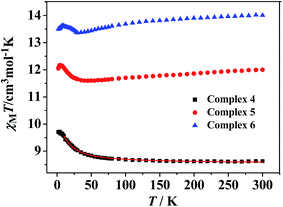
Static magnetic properties for 4–6. The temperature dependence of magnetic susceptibilities for 4–6 in the 2–300 K range is studied and shown in Fig. 6. For complex 4, the observed room temperature χMT value is 8.62 cm3 K mol−1, in agreement with the expected value 8.44 cm3 K mol−1 for one uncoupled GdIII ion (8S7/2, g = 2) and one organic radicals (S = 1/2). On decreasing the temperature, the χMT value steadily increases and begins to increase more sharply at 30 K till to reach a peak value of 9.72 cm3 K mol−1 at 2 K. The overall magnetic behavior indicates ferromagnetic interactions between the GdIII ion and nitroxide radicals. Accordingly, the system was modeled as a mononuclear bi-spin unit, and the magnetic analysis was carried out by using the spin Hamiltonian H = −2JRad-GdŜRadŜGd, where JRad-Gd characterized the exchange interactions for radical-Gd(III). Assuming that the radical and Gd(III) have the same g value, the magnetic data were analyzed by the following approximate treatment eqn (1) and (2). The mean-field approximation (zj′) was introduced to indicate the possible interactions between mononuclear moleculars.| |
 | (1) |
| |
 | (2) |
 |
| | Fig. 6 Temperature dependence of χMT values for complexes 4–6. The solid line represents the theoretical values based on the corresponding equations. | |
The observed χMT data were well reproduced (Fig. 6) by using the approximate eqn (1) and (2), giving the best fitting parameters of g = 2.02, JRad-Gd = 1.48 cm−1, zj′ = −0.014 cm−1. The positive value of JRad-Gd indicates the ferromagnetic interactions between Gd(III) and the radical, which is very common in the similar Gd(III)-radical complexes.19 In addition, the negative zj′ value indicates a very weak intermolecular antiferromagnetic interactions at low temperature.
The temperature dependence of magnetic susceptibility recorded for 5 and 6 revealed very similar behaviors (Fig. 6). At room temperature, the values of χMT are 11.97 and 14.02 cm3 K mol−1 for 5 and 6 mononuclear complexes, respectively. Both of the values are very close to the expected values of 12.12 and 14.54 cm3 K mol−1 for an uncoupled system of one Ln(III) ion (Tb(III) or Dy(III)) and one radical. When the temperature is lowered, the χMT values for 5 and 6 decrease slightly and reach values of 11.59 and 13.36 cm3 K mol−1 at about 40 K, respectively. Below this temperature, the χMT values increase slowly to the highest value of 12.21 cm3 K mol−1 at 6 K (for 5) and 13.62 cm3 K mol−1 at 7 K (for 6), and then decreases on further cooling. The decrease of χMT value upon lowering of the temperature in the high-temperature range for 5 and 6 is most probably governed by depopulation of the LnIII Stark sublevels. The increase of χMT at low temperature suggests the presence of weak ferromagnetic interaction between the Tb(III) or Dy(III) ion and the coordinated NO group of organic radical.
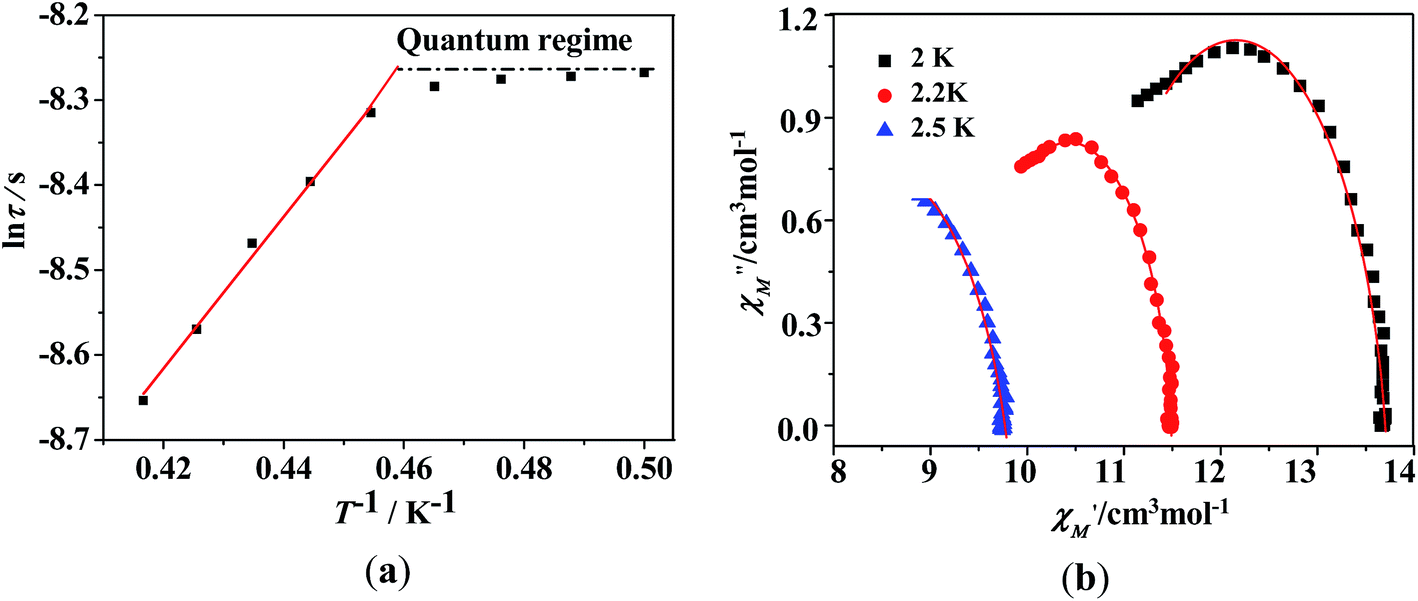
Dynamic magnetic properties for TbIII and DyIII's complexes (2, 3, 5, 6). TbIII and DyIII's complexes always have the tendency to be SMMs; therefore we performed the dynamic magnetic susceptibility measurements of complexes 2, 3, 5, 6.For complex 2, no out-of-phase signals were observed above 2 K as shown in Fig. S5 (see ESI†). The temperature and frequency dependency data of the alternating current susceptibilities for 3 under zero dc field (Fig. 7) show strong frequency and temperature dependencies. From the temperature dependencies of the ac susceptibilities (Fig. 7, left), the in-phase (χ′) signals show a maximum at frequencies above 1000 Hz, and the out-of-phase (χ′′) signals exhibit no maximum. From frequency dependencies of the ac susceptibility (Fig. 7, right), the magnetization relaxation times (τ) have been estimated between 2 and 2.5 K (Fig. 8a). Between 2.2–2.4 K, the relaxation follows a thermally activated mechanism affording an energy barrier of 6 K with a pre-exponential (τ0) of 4 × 10−6 s based on Arrhenius law [τ = τ0exp(Ueff/kBT)], which is consistent with those reported for similar SMMs (in the 10−6 to 10−11 s range).27,28 While at low temperatures a gradual crossover to a temperature-independent regime is observed. Below about 2.1 K, a dominant temperature-independent quantum regime of dynamics with a τ value of 0.00025 s explains the absence of the M versus H hysteresis effect at 2 K (Fig. 5b). This may due to the hyperfine couplings and dipolar spin–spin interactions in lanthanide ions, which allows fast quantum tunneling of magnetization that prevents the isolation of zero-field lanthanide SMMs with large barriers.5b,28b,29 From frequency dependencies of the ac susceptibility measurements, Cole–Cole diagrams in the form of χ′′ versus χ′ with nearly semicircular shapes have also been obtained (Fig. 8b). These data have been fitted to the generalized Debye model,30 giving the small distribution coefficient α value 0.13–0.09 (between 2–2.5 K), indication the narrow distribution of relaxation times at these temperatures. The frequency shift parameter φ is 0.16 (φ = (ΔTp/Tp)/Δ(logν)), excluding the possibility of spin-glass behavior.
 |
| | Fig. 7 Temperature dependence of the in-phase (top) and out-of-phase (bottom) components of the ac magnetic susceptibility for complex 3 under zero dc field at different frequencies (left). Frequency dependence of in-phase and out-of-phase susceptibilities under zero dc field at different temperatures for complex 3 (right). | |
 |
| | Fig. 8 (a) Magnetization relaxation time, lnτ vs. T−1 plot for 3 under zero-dc field. The solid line is fitted with the Arrhenius law. (b) Cole–Cole plots measured at 2–2.5 K under zero dc field for complex 3; the solid lines are the best fit to the experimental data. | |
For complex 5, frequency-dependent out-of-phase signals are observed (Fig. 9), indicating the onset of magnetization expected for single-molecule magnet (SMM) behavior. However, no peak maximum is found above 2 K even for the highest frequency investigated. This may result from quantum tunneling of the magnetization (QTM) that is too fast to be observed at the operating limits of our susceptometer. The imaginary component χ′′ of the complex 6 does not show any frequency-dependent phenomenon (Fig. S6, see ESI†).
 |
| | Fig. 9 Temperature dependence of the in-phase (χ′) and out-of-phase (χ′′) components of the ac magnetic susceptibility for complex 5 under zero dc field. | |
As we seen, by using Phtfac to replace hfac as β-diketonate, the crystal structures of complexes 5 and 6 show drastic changes compared with 2 and 3. In 2 and 3, two Ln(hfac)3 units are connected by a radical ligand MeTrzNIT to give binuclear cores, and the center LnIII ions are all surrounded with a slightly distorted 4,4,4-tricapped trigonal prism LnO8N coordination sphere. In 5 and 6, the central LnIII ions exhibit mononuclear structure, which are in an LnO7N coordination sphere with D4d symmetry. In addition, the presence of the different magnetic exchange coupling between the radical and metal ion in the two Tb's complexes or the two Dy's complexes will moderate their magnetic relaxation behaviors. It has been demonstrated that the ferromagnetic coupling between radical and 4f ions could enhance the aniostropy.22,31 Owning to the differences in crystal structure and magnetic interaction between the spin carries, there is also obvious change in their magnetic behavior: complex 2 shows no obvious magnetic relaxation whereas complex 5 exhibits SMM behavior. Complex 3 affords a barrier of 6 K while complex 6 has no visible magnetic relaxation. These results suggest that the local ligand-field of the Ln(III) ions and the magnetic coupling between the radical and Ln(III) ion are responsible for the different magnetic dynamic behaviors. It is evident that replacement of the CF3 group by a phenyl ring results in a significant change in magnetic relaxation and this provides an opportunity to fine-tune Ln-radical based SMM behavior through the modification of the β-diketonate coligand of lanthanide ion.
Conclusions
In summary, six novel binuclear or mono lanthanide-radical compounds have been synthesized using a triazole nitronyl nitroxide radical ligand and two different β-diketonate coligands. By using hexafluoroacetylacetone (hfac) as coligand, three binuclear tri-spin complexes were obtained, in which the nitronyl nitroxide moiety acts as a double-bridge ligand linking two Ln(III) ions by the two oxygen atoms of the N–O groups and the two nitrogen atoms of the triazole ring. When hfac were replaced by 4,4,4-trifluoro-1-phenylbutane-1,3-dione (Phtfac), three isomorphous mononuclear complexes 4–6 were obtained. The study of dynamics of the magnetizations for complexes 2, 3, 5, 6 shows that they exhibit quite distinct magnetic relaxation behaviors. Complex 3 and 5 shows frequency-dependent out-of-phase signals, however, such a phenomena is not observed for 2 and 6. The difference in magnetic relaxation of these complexes is probably due to the different symmetry of local ligand field of the Ln(III) (Tb and Dy) ions together with the different magnetic exchange coupling. These results show that the different ligand field can drastically affect the magnetic relaxation of the magnetization. Theoretical studies are required thoroughly analyze the symmetry of local ligand field of the Ln(III) ion/dynamic of the magnetization relationship.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21371133).
References
-
(a) R. Sessoli, D. Gatteschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS;
(b) G. Christou, D. Gatteschi, D. N. Hendrickson and R. Sessoli, MRS Bull., 2000, 25, 66 CrossRef CAS.
-
(a) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS;
(b) R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837 CrossRef;
(c) C. Coulon, H. Miyasaka and R. Clérac, Struct. Bonding, 2006, 122, 163 CrossRef CAS;
(d) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed;
(e) L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750 RSC.
-
(a) L. Thomas, F. Lionti, R. Ballou, D. Gatteschi, R. Sessoli and B. Barbara, Nature, 1996, 383, 145 CrossRef CAS;
(b) J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830 CrossRef CAS PubMed;
(c) J. A. Jones, Science, 1998, 280, 229 CrossRef CAS;
(d) W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406 CrossRef PubMed;
(e) B. Barbara, Inorg. Chim. Acta, 2008, 361, 3371 CrossRef CAS.
-
(a) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed;
(b) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed;
(c) M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, Ph. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417 CrossRef CAS PubMed.
-
(a) P. D. W. Boyd, Q. Li, J. B. Vincent, K. Folting, H. R. Chang, W. E. Streib, J. C. Huffman, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1988, 110, 8537 CrossRef CAS;
(b) N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., 2005, 117, 2991 (Angew. Chem., Int. Ed., 2005, 44, 2931) CrossRef;
(c) X. Y. Wang, Z. M. Wang and S. Gao, Chem. Commun., 2008, 44, 281 RSC.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 269 Search PubMed.
-
(a) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., 2001, 113, 1810 CrossRef;
(b) H. L. Sun, Z. M. Wang and S. Gao, Coord. Chem. Rev., 2010, 254, 1081 CrossRef CAS;
(c) J. S. Miller and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3065 RSC.
-
(a) J. K. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729 CrossRef CAS PubMed;
(b) K. Bernot, J. Luzon, L. Bogani, M. Etienne, C. Sangregorio, M. Shanmugam, A. Caneschi, R. Sessoli and D. Gatteschi, J. Am. Chem. Soc., 2009, 131, 5573 CrossRef CAS PubMed;
(c) K. Bernot, F. Pointillart, P. Rosa, M. Etienne, R. Sessoli and D. Gatteschia, Chem. Commun., 2010, 46, 6458 RSC.
-
(a) P. H. Lin, T. J. Burchell, R. Clérac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848 CrossRef CAS PubMed;
(b) G. F. Xu, Q. L. Wang, P. Gamez, Y. Ma, R. Clérac, J. K. Tang, S. P. Yan, P. Cheng and D. Z. Liao, Chem. Commun., 2010, 46, 1506 RSC;
(c) R. A. Layfield, J. J. W. McDouall, S. A. Sulway, F. Tuna, D. Collison and R. E. P. Winpenny, Chem.–Eur. J., 2010, 16, 4442 CrossRef CAS PubMed.
-
(a) I. J. Hewitt, Y. Lan, C. E. Anson, J. Luzon, R. Sessoli and A. K. Powell, Chem. Commun., 2009, 45, 6765 RSC;
(b) K. Isele, F. Gigon, A. F. Williams, G. Bernardinelli, P. Franz and S. Decurtins, Dalton Trans., 2007, 332 RSC;
(c) K. W. Galloway, A. M. Whyte, W. Wernsdorfer, J. Sanchez-Benitez, K. V. Kamenev, A. Parkin, R. D. Peacock and M. Murrie, Inorg. Chem., 2008, 47, 7438 CrossRef CAS PubMed;
(d) P. H. Lin, T. J. Burchell, L. Ungur, L. F. Chibotaru, W. Wernsdorfer and M. Murugesu, Angew. Chem., Int. Ed., 2009, 48, 9489 CrossRef CAS PubMed;
(e) Y. N. Guo, G. F. Xu, P. Gamez, L. Zhao, S. Y. Lin, R. P. Deng, J. K. Tang and H. J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538 CrossRef CAS PubMed.
- Y. N. Guo, G. F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H. J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948 CrossRef CAS PubMed.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS.
-
(a) N. Ishii, Y. Okamura, S. Chiba, T. Nogami and T. Ishida, J. Am. Chem. Soc., 2008, 130, 24 CrossRef CAS PubMed;
(b) C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369 CrossRef CAS PubMed;
(c) D. Luneau and P. Rey, Coord. Chem. Rev., 2005, 249, 2591 CrossRef CAS;
(d) R. N. Liu, L. C. Li, X. L. Wang, P. P. Yang, C. Wang, D. Z. Liao and J. P. Sutter, Chem. Commun., 2010, 46, 2566 RSC.
-
(a) K. Fegy, D. Luneau, T. Ohm, C. Paulsen and P. Rey, Angew. Chem., 1998, 110, 1331 (Angew. Chem., Int. Ed., 1998, 37, 1270) CrossRef;
(b) Y. Z. Zheng, Y. Lan, C. E. Anson and A. K. Powell, Inorg. Chem., 2008, 47, 10813 CrossRef CAS PubMed;
(c) S. Kanegawa, M. Maeyama, M. Nakano and N. Koga, J. Am. Chem. Soc., 2008, 130, 3079 CrossRef CAS PubMed;
(d) S. Katagiri, Y. Tsukahara, Y. Hasegawa and Y. Wada, Bull. Chem. Soc. Jpn., 2007, 80, 1492 CrossRef CAS.
- A. Lang, Y. Pei, L. Ouahab and O. Kahn, Adv. Mater., 1996, 8, 60 CrossRef CAS.
-
(a) G. M. Sheldrick, SHELXS-97, Program for Solution of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, SHELXL-97, Program for Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed;
(c) G. M. Sheldrick, SADABS, Program for Area Detector Adsorption Correction, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203 CrossRef.
-
(a) C. Benelli, A. Caneschi, D. Gatteschi, L. Pardi, P. Rey, D. P. Shum and R. L. Carlin, Inorg. Chem., 1989, 28, 272 CrossRef;
(b) J. P. Sutter, M. L. Kahn, S. Golhen, L. Ouahab and O. Kahn, Chem.–Eur. J., 1998, 4, 571 CrossRef CAS;
(c) Y. L. Wang, Y. Y. Gao, Y. Ma, Q. L. Wang, L. C. Li and D. Z. Liao, CrystEngComm, 2012, 14, 4706 RSC;
(d) C. Benelli, A. Caneschi, D. Gatteschi, J. Laugier and P. Rey, Angew. Chem., Int. Ed., 1987, 26, 913 CrossRef;
(e) C. Lescop, G. Bussiere, R. Beaulac, H. Beslisle, E. Beloeizky, P. Rey, C. Reber and D. Luneau, J. Phys. Chem. Solids, 2004, 65, 773 CrossRef CAS;
(f) N. Zhou, Y. Ma, C. Wang, G. F. Xu, J. K. Tang and D. Z. Liao, J. Solid State Chem., 2010, 183, 927 CrossRef CAS;
(g) Q. H. Zhao, Y. P. Ma, L. Du and R. B. Fang, Transition Met. Chem., 2006, 31, 593 CrossRef CAS.
- Y. Q. Sun, L. L. Fan, D. Z. Gao, Q. L. Wang, M. Du, D. Z. Liao and C. X. Zhang, Dalton Trans., 2010, 39, 9654 RSC.
- S. Y. Zhou, X. Li, T. Li, L. Tian, Z. Y. Liu and X. G. Wang, RSC Adv., 2015, 5, 17131 RSC.
- C. Aronica, G. Pilet, G. Chastanet, W. Wernsdorfer, J. F. Jacquot and D. Luneau, Angew. Chem., Int. Ed., 2006, 45, 4659 CrossRef CAS PubMed.
-
(a) J. X. Xu, Y. Ma, D. Z. Liao, G. F. Xu, J. K. Tang, C. Wang, N. Zhou, S. P. Yan, P. Cheng and L. C. Li, Inorg. Chem., 2009, 48, 8890 CrossRef CAS PubMed;
(b) P. Hu, X. F. Wang, Y. Ma, Q. L. Wang, L. C. Li and D. Z. Liao, Dalton Trans., 2014, 43, 2234 RSC.
-
(a) S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420 CrossRef CAS PubMed;
(b) K. Bernot, L. Bogani, R. Sessoli and D. Gatteschi, Inorg. Chim. Acta, 2007, 360, 3807 CrossRef CAS;
(c) Y. Ma, G. F. Xu, X. Yang, L. C. Li, J. K. Tang, S. P. Yan, P. Cheng and D. Z. Liao, Chem. Commun., 2010, 46, 8264 RSC;
(d) Y. M. Bing, N. Xu, W. Shi, K. Liu and P. Cheng, Chem.–Asian J., 2013, 8, 1412 CrossRef CAS PubMed.
-
(a) X. L. Mei, Y. Ma, L. C. Li and D. Z. Liao, Dalton Trans., 2012, 41, 505 RSC;
(b) X. L. Mei, R. N. Liu, C. Wang, P. P. Yang, L. C. Li and D. Z. Liao, Dalton Trans., 2012, 41, 2904 RSC.
-
(a) N. Zhou, Y. Ma, C. Wang, G. F. Xu, J. K. Tang, J. X. Xu, S. P. Yan, P. Cheng, L. C. Li and D. Z. Liao, Dalton Trans., 2009, 38, 8489 RSC;
(b) T. Han, W. Shi, X. P. Zhang, L. L. Li and P. Cheng, Inorg. Chem., 2012, 51, 13009 CrossRef CAS PubMed.
-
(a) S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448 CrossRef CAS PubMed;
(b) G. J. Chen, C. Y. Gao, J. L. Tian, J. K. Tang, W. Gu, X. Liu, S. P. Yan, D. Z. Liao and P. Cheng, Dalton Trans., 2011, 40, 5579 RSC;
(c) G. J. Chen, Y. N. Guo, J. L. Tian, J. K. Tang, W. Gu, X. Liu, S. P. Yan, P. Cheng and D. Z. Liao, Chem.–Eur. J., 2012, 18, 2484 CrossRef CAS PubMed;
(d) Y. L. Wang, Y. Ma, X. Yang, J. K. Tang, P. Cheng, Q. L. Wang, L. C. Li and D. Z. Liao, Inorg. Chem., 2013, 52, 7380 CrossRef CAS PubMed;
(e) H. X. Tian, R. N. Liu, X. L. Wang, P. P. Yang, Z. X. Li, L. C. Li and D. Z. Liao, Eur. J. Inorg. Chem., 2009, 4498 CrossRef CAS;
(f) R. N. Liu, C. M. Zhang, L. C. Li, D. Z. Liao and J.-P. Sutter, Dalton Trans., 2012, 41, 12139 RSC;
(g) F. Pointillart, K. Bernot, G. Poneti and R. Sessoli, Inorg. Chem., 2012, 51, 12218 CrossRef CAS PubMed.
-
(a) Y. Wang, X. L. Li, T. W. Wang, Y. Song and X. Z. You, Inorg. Chem., 2010, 49, 969 CrossRef CAS PubMed;
(b) D. P. Li, T. W. Wang, C. H. Li, D. S. Liu, Y. Z. Li and X. Z. You, Chem. Commun., 2010, 46, 2929 RSC;
(c) J. Liu, X. P. Zhang, T. Wu, B. B. Ma, T. W. Wang, C. H. Li, Y. Z. Li and X. Z. You, Inorg. Chem., 2012, 51, 8649 CrossRef CAS PubMed.
-
(a) F. Luis, M. Martínez-Peérez, O. Montero, E. Coronado, S. Cardona-Serra, C. Martí-Gastaldo, J. M. Clemente-Juan, J. Seseé, D. Drung and T. Schurig, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 060403 CrossRef;
(b) R. Giraud, W. Wernsdorfer, A. M. Tkachuk, D. Mailly and B. Barbara, Phys. Rev. Lett., 2001, 87, 057203 CrossRef CAS PubMed.
-
(a) K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS;
(b) S. M. J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L. C. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329 CrossRef CAS.
- J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.