
Novel magnetically separable ZnO/AgBr/Fe3O4/Ag3VO4 nanocomposites with tandem n–n heterojunctions as highly efficient visible-light-driven photocatalysts
Abstract
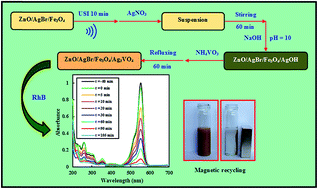
In the present work, novel magnetically separable ZnO/AgBr/Fe3O4/Ag3VO4 nanocomposites with different weight percentages of Ag3VO4 were successfully prepared. The preparation method is a facile large-scale method with a low temperature of 96 °C. Characterization techniques of XRD, EDX, SEM, TEM, UV-vis DRS, FT-IR, PL, and VSM were applied to study the microstructure, purity, morphology, spectroscopic, and magnetic properties of the resultant samples. Photocatalytic activity was evaluated by degradation of rhodamine B under visible-light irradiation. The results indicated that the nanocomposite with 10% Ag3VO4 has the best activity. The photocatalytic activity of this nanocomposite is about 2.8 and 3.5-fold higher than those of the ZnO/AgBr/Fe3O4 and ZnO/Ag3VO4/Fe3O4 nanocomposites, respectively. The highly enhanced activity was attributed to the formation of tandem n–n heterojunctions between ZnO, AgBr, and Ag3VO4 which suppresses recombination of the photogenerated electron–hole pairs. Furthermore, the enhanced activity of the nanocomposite for degradation of two more dye pollutants and a colorless pollutant was confirmed. Finally, the magnetic photocatalyst was recycled using an external magnetic field with reasonable activity after four runs.
 Please wait while we load your content...
Please wait while we load your content...

