
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radiosynthesis and characterisation of a potent and selective GPR139 agonist radioligand†
Sebastiaan
Kuhne‡
ab,
Anne Cathrine
Nøhr‡
a,
Aleš
Marek
c,
Tomáš
Elbert
c,
Anders Bue
Klein
a,
Hans
Bräuner-Osborne
a,
Petrine
Wellendorph
 a and
Daniel Sejer
Pedersen
*a
a and
Daniel Sejer
Pedersen
*a
aDepartment of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark. E-mail: daniel.pedersen@sund.ku.dk
bAmsterdam Institute for Molecules, Medicines and Systems (AIMMS), Division of Medicinal Chemistry, Faculty of Science, VU University Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands
cInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, 16610, Prague 6, Czech Republic
First published on 17th December 2015
Abstract
Compound 1 is a selective and potent agonist of the G protein-coupled receptor GPR139 (EC50 = 39 nM). In this study, we describe the synthesis, radiolabelling and in vitro evaluation of [3H]-1 for the characterisation of GPR139 and its spatial expression in the brain using autoradiography. Two different synthesis routes for the radiolabelling of 1 based on a reductive debromination strategy were investigated using deuterium (D2, g). The route based on reductive debromination of the bromonaphthyl precursor 5 proved superior over arylbromide 4 and was employed for the radiolabelling experiments. Reductive debromination of precursor 5 was accomplished using 3H2, Pd/C and triethylamine in DMF at ambient temperature to give target molecule [3H]-1 with a specific activity of 19.3 Ci mmol−1 and a radiochemical purity of ≥95%. By application of autoradiography and binding studies, it was not possible to discriminate [3H]-1 binding to wildtype mice brains from GPR139 knockout mice brains and total binding from non-specific binding in CHO-k1 cells stably expressing human GPR139 receptor. Based on these experiments we conclude that [3H]-1 is not a suitable radioligand for the characterisation of GPR139.
Introduction
G protein-coupled receptors (GPCRs) constitute one of the largest protein families in the human proteome,1 targeted by approximately 30% of currently marketed drugs.2 GPCRs have been the focus of extensive studies for decades, but still many GPCRs remain for which the endogenous ligands are unknown (orphan receptors). The unravelling and characterisation of orphan receptors is of utmost importance to determine their physiological role and evaluate their potential as targets for drug development. GPR139 is an orphan class A GPCR.3 GPR139 mRNA is exclusively expressed in the CNS in particular in striatum and hypothalamus, and it has been suggested that GPR139 plays a role in the modulation of locomotor activity or food intake.4–6 Several ligands with different chemotypes have been reported for GPR139 (Fig. 1). A screening campaign by Shi et al.7 followed up by a structure-activity study identified 1 as a potent GPR139 agonist. In addition, an agonist (LP-360924) and two antagonists (LP-471756 and LP-114958) were identified in a high-throughput screen by Hu et al.8 Recently, we have suggested that L-tryptophan (L-Trp) and L-phenylalanine (L-Phe) may be the endogenous agonists for GPR139,9 using a pharmacophore model based on the ligands described by Shi et al.7 During the preparation of this manuscript, scientists at Janssen R&D confirmed our hypothesis that L-Phe and L-Trp are potential endogenous agonists and a surrogate agonist JNJ-63533054 and other analogues were disclosed.4,10 Moreover, Wang et al. recently reported five antagonists obtained from a screening of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds.11
000 compounds.11
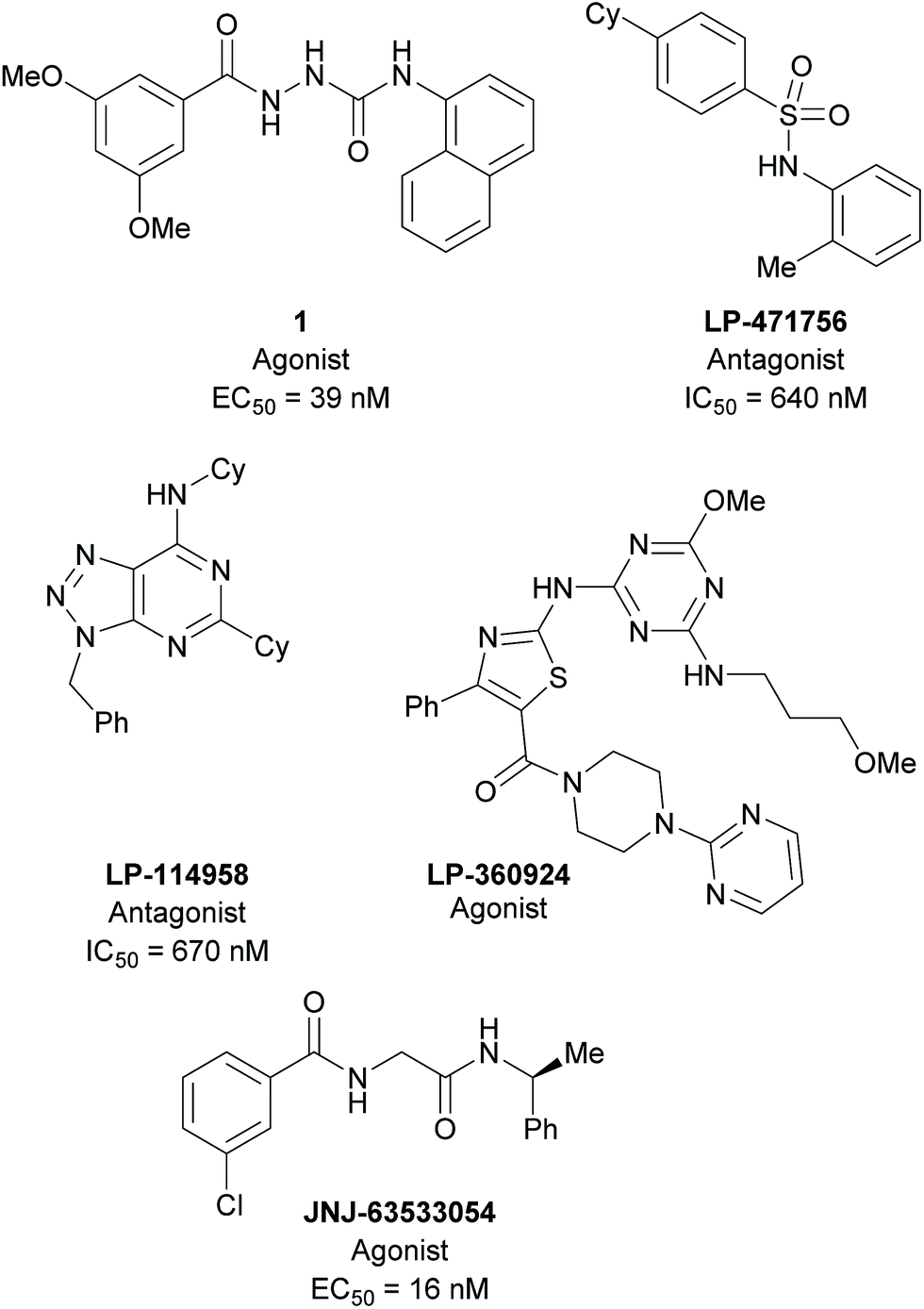 | ||
| Fig. 1 Selected ligands targeting GPR139. 1,7 LP-360924, LP-471756, LP-114958,8 and JNJ-63533054.4,10 | ||
In an effort to further characterise GPR139 and to determine its anatomical distribution a suitable radioligand was required. Binding experiments with a radioligand together with site-directed mutagenesis studies can be used to reveal key amino acid residues involved in ligand binding, identify novel receptor ligands and to determine binding kinetics. Moreover, autoradiography studies can reveal the spatial expression of proteins (e.g. GPR139) in the brain, which can give indications of receptor physiology and function. Ligand 1 is a potent GPR139 agonist that has been screened against a panel of 90 different receptors, enzymes, transporters and ion channels with no observed cross reactivity.7 Based on the high potency and in particular, excellent selectivity, 1 was selected as our candidate for development of a GPR139 radioligand.
Results and discussion
Ligand synthesis and radiosynthesis
Two routes based on a reductive debromination strategy starting from commercially available bromo-compounds 2 and 3 were considered for the synthesis of a radioligand (Scheme 1).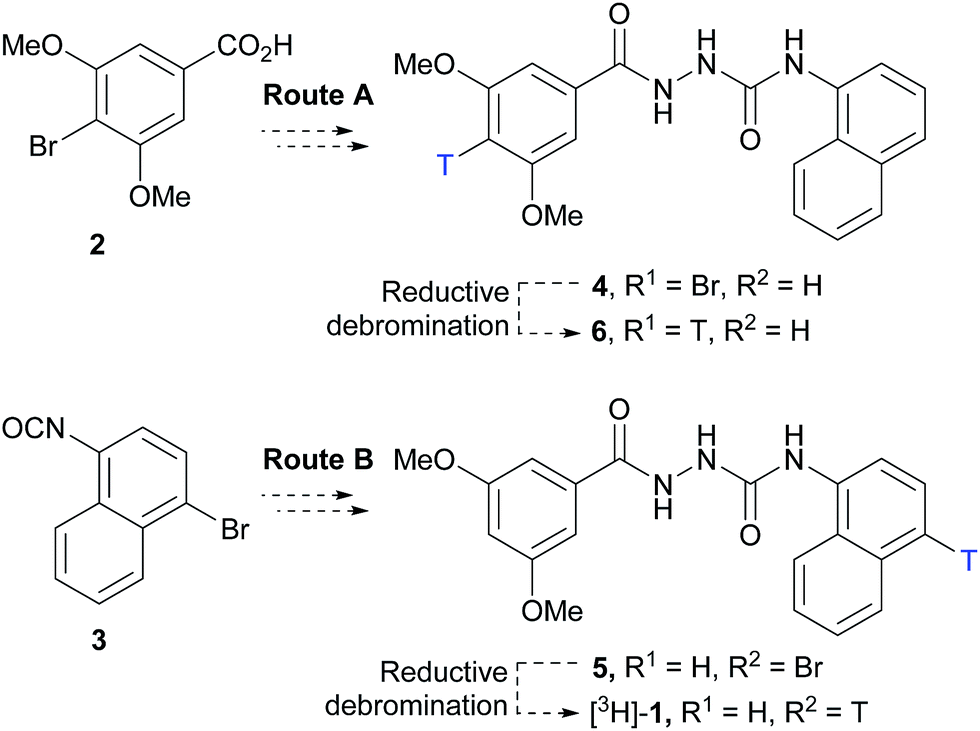 | ||
| Scheme 1 Synthetic routes considered for the synthesis of a GPR139 radioligand. Synthesis of mono-bromo precursors 4 and 5 followed by reductive debromination to give mono-tritiated radioligands 6 and [3H]-1, respectively. | ||
We set out to evaluate both reductive debromination routes initially using deuterium gas in the reductive debromination step, to identify the route best suited to proceed to radiolabelling (Scheme 1). The two substrates 4 and 5 for reductive debromination and the non-labelled agonist 1 were synthesised as outlined in Schemes 2 and 3. Carboxylic acid 2 was converted to methyl ester 7 using sulfuric acid in methanol followed by reaction with hydrazine hydrate in ethanol to provide hydrazide 8. Subsequent coupling of hydrazide 8 with 1-isocyanatonaphthalene gave substrate 4 in good overall yield, ready for reductive debromination (Scheme 2). Analogously, substrate 5 and agonist 1 were synthesised in good overall yield (Scheme 3). It should be noted that due to the poor solubility of the hydrazides in THF it was necessary to add methanol to facilitate the reaction with the isocyanates.
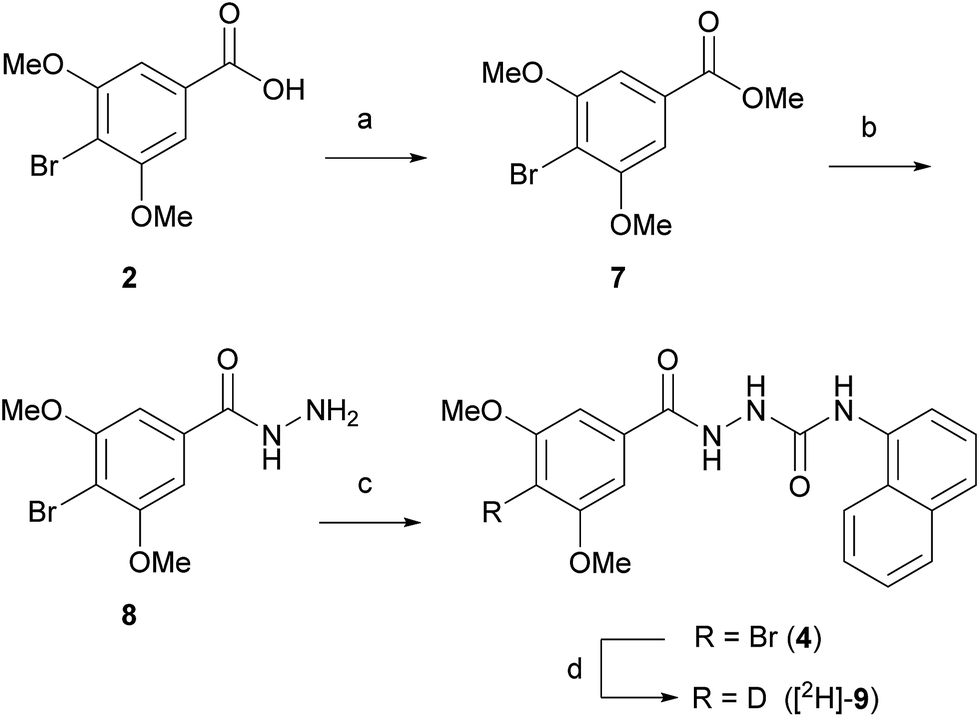 | ||
| Scheme 2 Reagents and conditions: (a) H2SO4, MeOH, reflux, 16 h, 84%; (b) hydrazine hydrate, EtOH, 50 °C, 8 h, 57%; (c) 1-isocyanatonaphthalene, THF, MeOH, 1 h, 78%; (d) D2, Pd/C, DMF, rt, 30.5 h, 36% (ratio [2H]-9:1 – 4:1). | ||
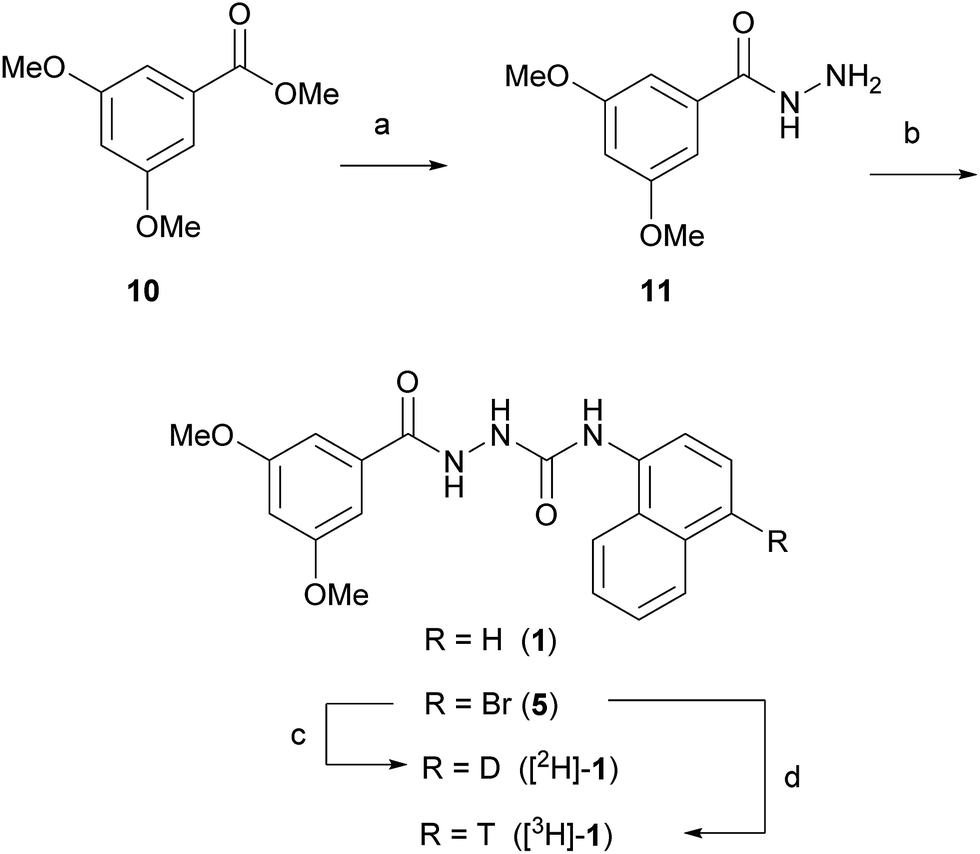 | ||
| Scheme 3 Reagents and conditions: (a) hydrazine hydrate, EtOH, 55 °C, 8 h, 32%; (b) 1: 1-isocyanatonaphthalene, THF, MeOH, 1.5 h, 70%; 5: 1-bromo-4-isocyanatonaphthalene, THF, MeOH, 1 h, 81%; (c) D2, Pd/C, Et3N, DMF, 1 h, >80% (ratio [2H]-1:1 – 4:1); (d) T2, Pd/C, Et3N, DMF, 1 h, (ratio [3H]-1:1 – 5:2). | ||
With substrates 4 and 5 in hand, we set out to evaluate these in the final reductive debromination step. Two critical parameters were considered for the reductive debromination, namely the formation of undesired side products, and the reaction rate. Due to the poor solubility of substrates 4 and 5, it was necessary to perform the reductive debromination step in DMF. The initial reductive debromination reactions were performed on a 30 mg scale under identical conditions. Deuteration of substrate 4 using deuterium gas and palladium on carbon gave a mixture of compounds [2H]-9 and 1 in a ∼4:1 ratio, whereas substrate 5 gave a mixture of compound [2H]-1 and 1 in a ∼1:1 ratio (determined by 1H NMR). The formation of side products during both reductive debromination reactions was minor and comparable according to HPLC (see ESI† for details). Although deuterium incorporation was more efficient for substrate 4, the reaction time to obtain >95% conversion was much longer than for substrate 5 (>21 vs. 1 hour(s), respectively), thus the route employing substrate 5 was favoured. To confirm the result the reductive debromination of substrate 5 was reproduced on a 100 mg scale to give >93% conversion in approx. 1 hour ([2H]-1:1 ratio – 2:3, see ESI† for details). To further optimise the route for radiolabelling, triethylamine (3 eq.) was added to sequester HBr formed during the course of the reaction. Moreover, because the reaction would be run at higher dilution during radiosynthesis, the catalyst loading was increased substantially. This resulted in a significant increase in deuterium incorporation to produce a ∼4:1 mixture of [2H]-1vs.1, while maintaining a reaction time of approx. 1 hour, and was judged suitable for radiolabelling (see ESI† for details). Thus, substrate 5 was subjected to reductive debromination using tritium gas, triethylamine and palladium on carbon at room temperature for 1 hour, followed by purification by preparative HPLC (Fig. 2).
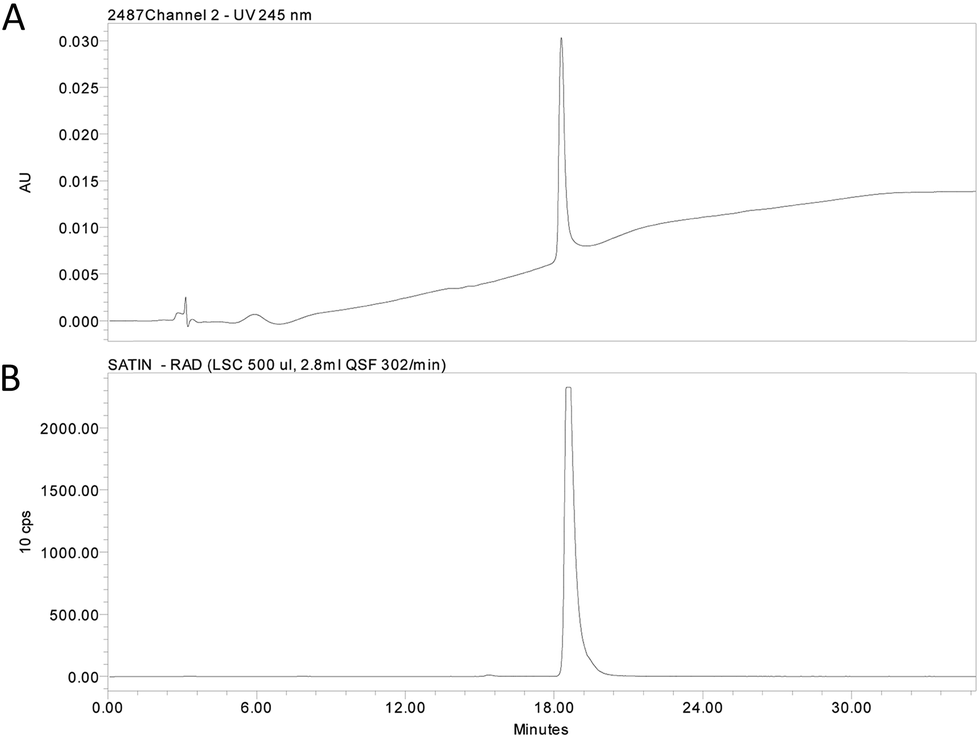 | ||
| Fig. 2 (A) HPLC chromatogram and (B) radio trace of the purified radioligand [3H]-1. | ||
In Fig. 3A, the 1H NMR spectra for the aromatic region of compounds 1, [2H]-1 and [3H]-1 are depicted. Tritium incorporation for [3H]-1vs.1 was estimated to be ∼5:2 based on 1H NMR (Fig. 3A and B) with a purity and radiochemical purity of ≥95% and a specific activity of 19.3 Ci mmol−1 (see ESI† for details).
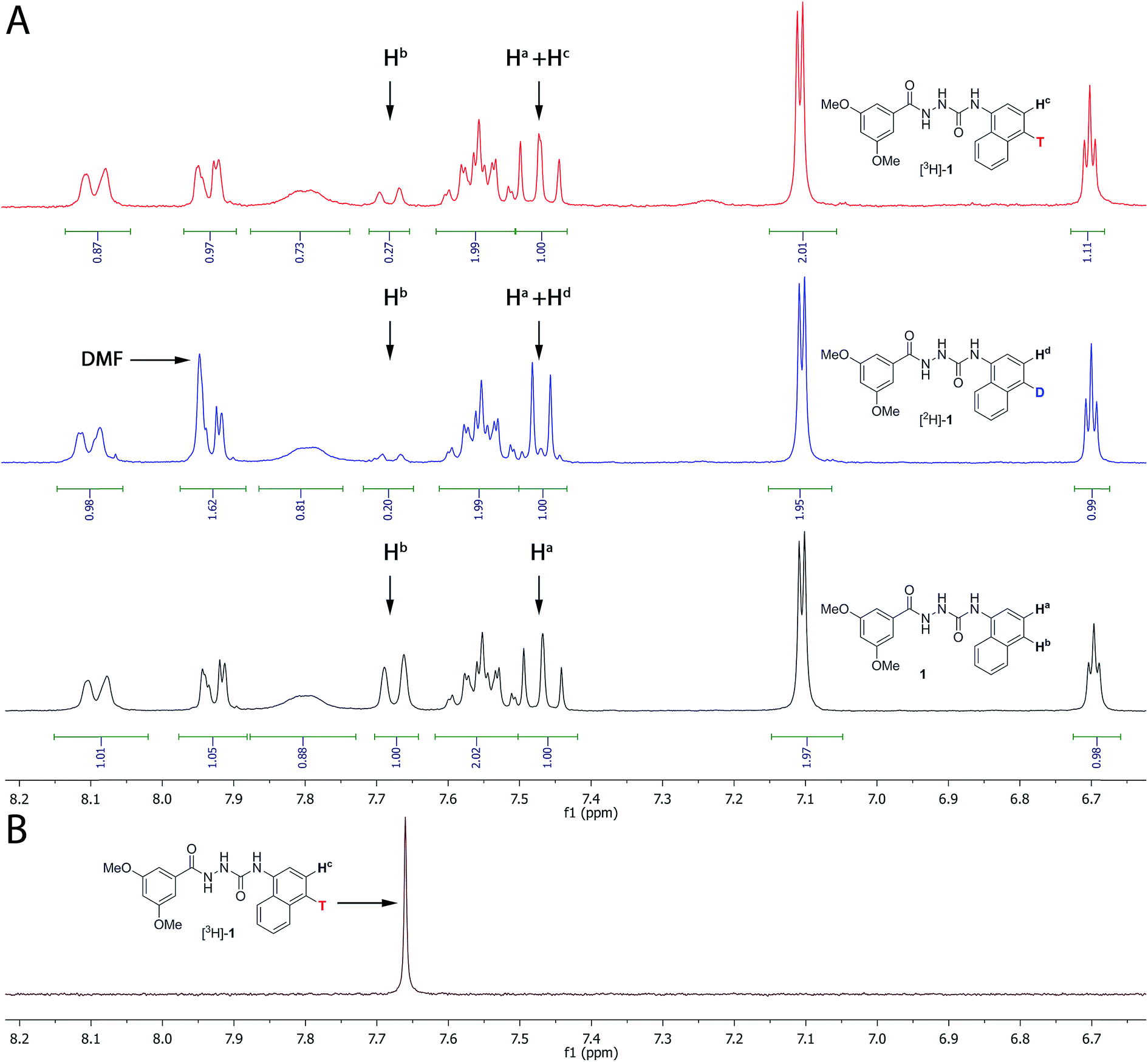 | ||
| Fig. 3 (A) 1H NMR of compound 1, [2H]-1 and [3H]-1 (only the aromatic region is shown, see ESI† for the full spectrum). Both the deuterated and tritiated product contain unlabelled compound 1. The ratio between labelled and unlabelled compound was determined by 1H NMR by using the integrals for the resonances at 7.67 ppm (corresponding to Hb in compound 1 only) and at 7.47 ppm (corresponding to Ha both in the labelled and unlabelled compounds). (B) Proton decoupled 3H NMR of [3H]-1. | ||
[3H]-1 was formulated in absolute ethanol at a radioactive concentration of 1 mCi ml−1 and the stability during storage was evaluated by HPLC. [3H]-1 proved to be very stable at −196 °C (2 and 10 months after formulation the radiopurity was determined to ≥94% and ≥90%, respectively). In contrast, when stored at −21 °C, decomposition was observed (99% radiopurity at day 0, 92% at day 26, 81% at 74 and 50% at day 321, see ESI† for details).
Native expression of GPR139
To determine the spatial expression of GPR139 in mouse brain, the distribution of [3H]-1 binding was assessed qualitatively by autoradiography in brain sections from 4.5 months old wildtype (WT) and GPR139 knockout (KO) mice (see ESI† for details). As illustrated in Fig. 4A and B, it is evident that [3H]-1 labels both the brain sections from the WT and GPR139 KO mice with the same intensity. Furthermore, the [3H]-1 binding was not significantly displaced by 10 μM unlabelled 1. The autoradiography was repeated under slightly modified conditions using a HEPES-buffer (20 mM HEPES, 1 mM MgCl2 and 1 mM CaCl2, 0.01% Pluronic, pH 7.4) with a similar result indicating that [3H]-1 has a high degree of non-specific binding.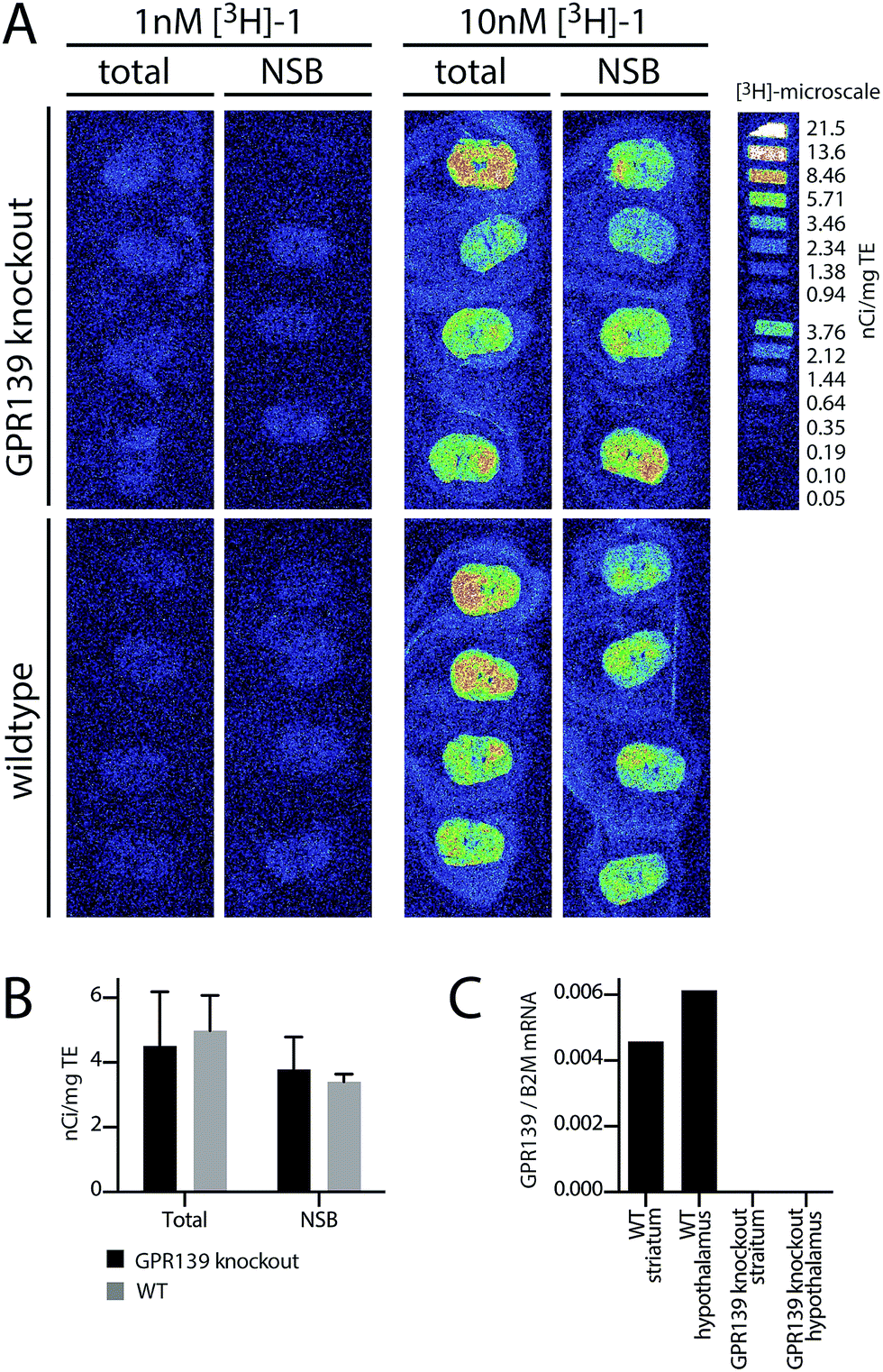 | ||
| Fig. 4 (A) Autoradiograms showing [3H]-1 binding in WT and GPR139 KO mice brains. The coronal brain sections were incubated with 1 nM or 10 nM [3H]-1 either in the absence (total) or presence (non-specific binding, NSB) of 10 μM 1. To the right [3H]-microscale in nCi mg−1 tissue equivalent is depicted. (B) Mean ± SD of mean nCi mg−1 tissue equivalent of the whole brain area. (C) qPCR analysis of GPR139 mRNA expression in the striatum and hypothalamus of WT and GPR139 KO mice. Levels are shown relative to the housekeeping gene B2M. | ||
To verify the presence of GPR139 in the striatum and hypothalamus,4–6 quantitative PCR (qPCR) was performed for both regions on both GPR139 WT and KO mice (see ESI† for details). Indeed, high levels of GPR139 mRNA could be detected in both brain regions of the WT mice, but not in KO mice (Fig. 4C).
Recombinant GPR139 CHO-k1 cell line
Next, we investigated the binding of [3H]-1 to the CHO-k1 GPR139 cell line (expressing the human GPR139) in a standard membrane filter-binding assay. Membrane binding to either CHO-k1 GPR139 or background CHO-k1 cells was performed with 5 nM or 50 nM [3H]-1 in a standard 50 mM Tris buffer, pH 7.4 (Fig. 5A, see ESI† for details). No difference in the binding of [3H]-1 to CHO-k1 GPR139 and CHO-k1 membranes was observed. Furthermore [3H]-1 binding was not displaced by 4 μM unlabelled 1. The filter membrane binding was repeated with slight modifications using two alternative buffers; (A) 50 mM Tris, 2.5 mM CaCl2, pH 7.4; and (B) 20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, pH 7.4; with similar results. Additionally, filter binding was assessed by pre-incubation of the GF/C unifilters with 0.2% bovine serum albumin or polyethylenimine, however all conditions resulted in high [3H]-1 binding to the filters (see ESI Fig. S1†).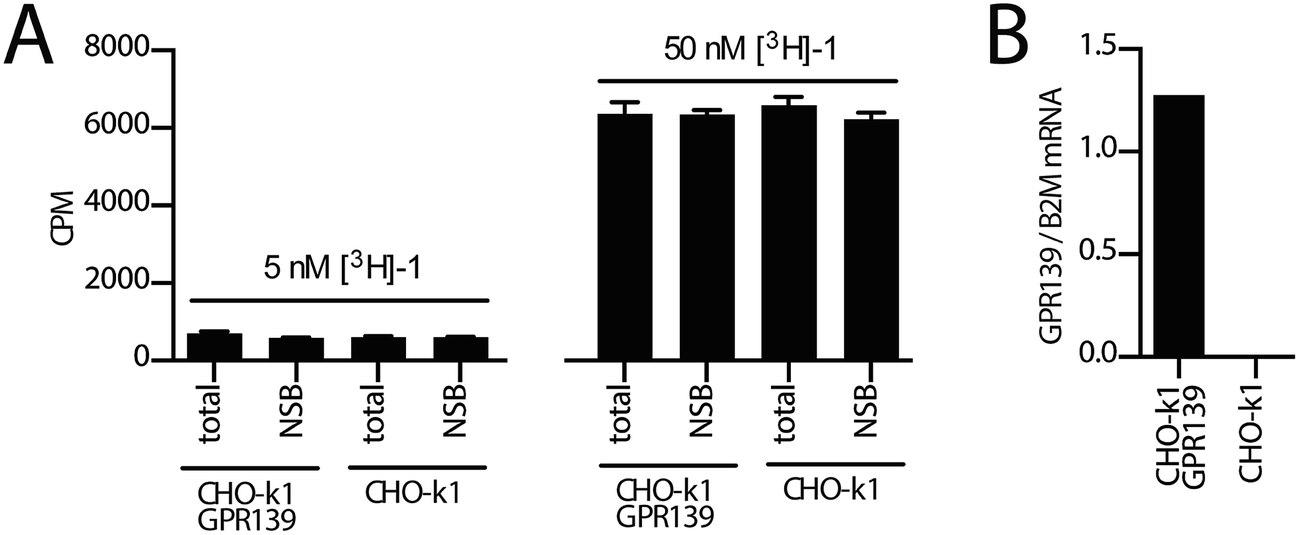 | ||
| Fig. 5 (A) [3H]-1 binding to membranes prepared from CHO-k1 GPR139 and CHO-k1 cell lines. Membranes were incubated with 5 nM or 50 nM [3H]-1 in either the absence (total) or presence (non-specific binding, NSB) of 4 μM unlabelled 1 in a 50 mM Tris buffer, pH 7.4. (B) qPCR analysis showing GPR139 mRNA levels relative to the reference gene B2M (beta-2 microglobulin) in the CHO-k1 GPR139 stable cell line and CHO-k1 cell line. | ||
To avoid the high level of binding to the filters and potential low specific binding due to a fast off-rate of the radioligand; SPA-bead binding, centrifugation-binding and whole cell binding was attempted, but none of these methods gave any significant difference in [3H]-1 binding between the total and non-specific binding (data not shown).
GPR139 is expressed at the protein level in the CHO-k1 GPR139 cell line, as we have previously demonstrated a functional agonist response to 1 in the same cell line,9 which was confirmed in the present study. Using qPCR analysis we furthermore verified the presence of GPR139 transcripts in the CHO-k1 GPR139 cell line but not in the CHO-k1 background cell line (Fig. 5B, see ESI† for details).
Thus, using a variety of radioligand binding assays, we show that [3H]-1 is not a suitable radioligand for detecting GPR139 in brain or in CHO-k1 GPR139 cell membranes. This might be due to the lipophilic character of 1 causing significant non-specific binding, low affinity or a high off-rate. The former is the most likely explanation as in particular the SPA and centrifugation protocols are performed in equilibrium and thus allow for detection of radioligands with relatively low affinity and higher off-rates.12 It is also possible that the GPR139 receptor protein level in the CHO-k1 cells are too low to allow for a sufficient level of specific binding, as it is not possible to determine GPR139 receptor levels quantitatively from the measured functional responses.
Compound 1 has previously been shown to be metabolically stable in human and rat liver microsomes.7 10 μM compound 1 shows no activity on native CHO-k1 cells and does not change the morphology of living CHO-k1 cells during ligand incubation (data not shown). Moreover, no cross-reactivity of compound 1 was observed against a broad Cerep panel of 90 receptors, ion channels and enzymes.7 Collectively, these data show that the lack of specific radioligand binding was neither due to metabolic instability of compound 1 or a result of low specific binding to the GPR139 receptor.
During the preparation of this manuscript, scientists at Janssen R&D reported synthesis and initial characterization of JNJ-63533054 as a tritium-labelled radioligand.4,10 This radioligand shows specific binding with a Kd of 10 nM in a GPR139-inducible Trex-CHO cell line in a membrane filter-binding assay. A comparison of clogP and logS for JNJ-63533054 vs. [3H]-1 resulted in similar values (clogP: 3.21 vs. 3.40, and logS: −4.53 vs. −5.46. Calculated with MOE 2014.0901)13 and thus does not provide an explanation for the negative results obtained with [3H]-1. Alternatively, the induced Trex-CHO cell line has higher expression of GPR139 than the stable CHO-k1 cell line used in this study, given the potential receptor down-regulation mediated by the L-Trp and L-Phe present in the cell culture medium during the cell culturing.4,9 However, we were not able to detect specific 1 binding to native receptors in brain slices with autoradiography. This could potentially also be explained by low receptor expression levels in the discrete brain regions showing mRNA expression as this has so far not been determined.
Conclusion
Radioligand [3H]-1 was successfully synthesised using a reductive debromination route. A variety of binding experiments with mouse brain tissue and cells recombinantly expressing GPR139 failed to show specific receptor binding, due to a high degree of non-specific binding of [3H]-1. Collectively, our data demonstrates that [3H]-1 is not suitable as a GPR139-specific radioligand.Acknowledgements
H. Lundbeck A/S is thanked for providing compound 1, the CHO-k1 GPR139 and CHO-k1 cell line. The Lundbeck Foundation and Hørslev Foundation are gratefully acknowledged for financial support. The authors thank the Academy of Sciences of the Czech Republic for financial support on this project (RVO: 61388963). S. K. was supported by the European Cooperation in Science and Technology (COST) Action CM1207: GPCR-Ligand Interactions, Structures, and Transmembrane Signalling: a European Research Network (GLISTEN), as well as the European University Consortium for Pharmaceutical Sciences (ULLA).References
- R. Fredriksson, M. C. Lagerstrom, L. G. Lundin and H. B. Schioth, Mol. Pharmacol., 2003, 63, 1256–1272 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- D. E. Gloriam, H. B. Schioth and R. Fredriksson, Biochim. Biophys. Acta, 2005, 1722, 235–246 CrossRef CAS PubMed.
- C. Liu, P. Bonaventure, G. Lee, D. Nepomuceno, C. Kuei, J. Wu, Q. Li, V. Joseph, S. Sutton, W. Eckert, X. Yao, L. Yieh, C. Dvorak, N. Carruthers, H. Coate, S. Yun, C. Dugovic, A. Harrington and T. Lovenberg, Mol. Pharmacol., 2015, 88(5), 911–925 CrossRef CAS PubMed.
- A. Matsuo, S. Matsumoto, M. Nagano, K. H. Masumoto, J. Takasaki, M. Matsumoto, M. Kobori, M. Katoh and Y. Shigeyoshi, Biochem. Biophys. Res. Commun., 2005, 331, 363–369 CrossRef CAS PubMed.
- U. Susens, I. Hermans-Borgmeyer, J. Urny and H. C. Schaller, Neuropharmacology, 2006, 50, 512–520 CrossRef PubMed.
- F. Shi, J. K. Shen, D. Chen, K. Fog, K. Thirstrup, M. Hentzer, J. J. Karlsson, V. Menon, K. A. Jones, K. E. Smith and G. Smith, ACS Med. Chem. Lett., 2011, 2, 303–306 CrossRef CAS PubMed.
- L. A. Hu, P. M. Tang, N. K. Eslahi, T. Zhou, J. Barbosa and Q. Liu, J. Biomol. Screening, 2009, 14, 789–797 CrossRef CAS PubMed.
- V. Isberg, K. B. Andersen, C. Bisig, G. P. Dietz, H. Bräuner-Osborne and D. E. Gloriam, J. Chem. Inf. Model., 2014, 54, 1553–1557 CrossRef CAS PubMed.
- C. A. Dvorak, H. Coate, D. Nepomuceno, M. Wennerholm, C. Kuei, B. Lord, D. Woody, P. Bonaventure, C. Liu, T. Lovenberg and N. I. Carruthers, ACS Med. Chem. Lett., 2015, 6, 1015–1018 CrossRef CAS PubMed.
- J. Wang, L. Y. Zhu, Q. Liu, M. Hentzer, G. P. Smith and M. W. Wang, Acta Pharmacol. Sin., 2015, 36, 874–878 CrossRef CAS PubMed.
- J. F. Glickman, A. Schmid and S. Ferrand, Assay Drug Dev. Technol., 2008, 6, 433–455 CrossRef CAS PubMed.
- MOE, version 2014.0901, Chemical Computing Group (CCG), Montreal, Canada, http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Reaction progress of reductive debromination reactions based on HPLC. Synthesis and characterisation of all compounds. Detailed description of the pharmacological assays, clogP and logS calculations. See DOI: 10.1039/c5ra21326f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |