
A chitosan–Pt nanoparticles/carbon nanotubes-doped phosphomolybdate nanocomposite as a platform for the sensitive detection of nitrite in tap water†
Abstract
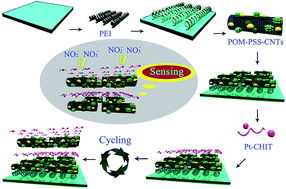
A composite multilayer film was fabricated based on H7P2Mo17V1O62 (P2Mo17V)–carbon nanotubes (CNTs) and Pt–chitosan (Pt–CHIT) nanoparticles using layer-by-layer self-assembly method. The incorporation of CNTs and Pt–CHIT nanoparticles into the P2Mo17V-based composite film endowed the modified electrode with a fast electron transfer rate and high electrocatalytic activity towards nitrite oxidation. The as-prepared sensor showed a fast response to nitrite with a good linear concentration range from 1.25 × 10−7 M to 4.167 × 10−3 M and a high sensitivity of 0.019 μA mM−1. Most noteworthy is its ultra-low detection limit of 3.8 × 10−9 M at the signal-to-noise ratio of 3 under optimal experimental conditions, which was below the detection limit of the vast majority of nitrite sensors reported before. The as-prepared sensor was able to distinguish the oxidation response of common interferences in solution mixtures under multiple potentials. It was successfully applied for determination of nitrite in tap water samples with satisfactory results.
 Please wait while we load your content...
Please wait while we load your content...

