
Characterization of water state and distribution in fibre materials by low-field nuclear magnetic resonance
Abstract
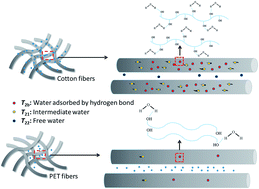
The water state and distribution in PET and cotton fibres were studied by low-field proton nuclear magnetic resonance. The spin–spin relaxation times (T2) were measured with single pulse free induction decay (FID). There are three different states of adsorbed water in the fibre materials. The slowest fraction T22 can be assigned to the bulk water. The intermediate component T21 can be ascribed to microporous structure confined water. The fastest fraction, T2b, can be assigned to the water molecules trapped by hydrogen bond owing to the chemical group. During desorption process of fibre materials, three types of water in the fibre materials work together to present the time-domain spectra, where three peaks are really related each other. Based on the interaction relationship between multi-structure of fibre materials and adsorbed water, PET fibre materials were chosen to investigate the adsorption and desorption behavior designed by copolymerization and morphology design method. The experiments of surface contact angle of fibre and fabric, moisture adsorption, water adsorption, wicking distance and water vapor permeability were carried out. The results show that the designed PET fibre materials have fast adsorption–desorption capacity. LF-NMR provides unique insight into the water state and distribution of multi-structure of fibre materials.
 Please wait while we load your content...
Please wait while we load your content...

