
First principles study of the adsorption and dissociation mechanisms of H2S on a TiO2 anatase (001) surface†
Naeem Shahzad‡
ac,
Akhtar Hussainbcd,
Naeem Mustafab,
Nisar Alie,
Mohammed Benali Kanounf and
Souraya Goumri-Said*f
aNational University of Sciences and Technology, Islamabad, Pakistan
bTPD, Pakistan Institute of Nuclear Science and Technology (PINSTECH), P. O. Nilore, Islamabad, Pakistan. E-mail: ahmohal@yahoo.com
cNational Centre for Physics, Quaid-e-Azam University Campus, Islamabad, Pakistan
dNED, Pakistan Institute of Engineering and Applied Sciences (PIEAS), P. O. Nilore, Islamabad, Pakistan
eDepartment of Physics, GPG Jehanzeb College Swat, Pakistan
fCollege of Science, Physics department, Al Faisal University, Riyadh 11533, Saudi Arabia. E-mail: souraya.goumri-said@chemistry.gatech.edu; sosaid@alfaisal.edu; mkanoun@alfaisal.edu
First published on 28th December 2015
Abstract
The adsorption and dissociation mechanisms of H2S on a TiO2 (001) surface were elucidated using first principles calculation based on the density functional theory. The interaction of the intermediates involving S, HS and OH species has been discussed in detail. For these species, the most favorable adsorption sites were determined and led to further computations involving dissociation of H2S into HS and H and bonding of H-atom with the OH to form water on the surface. The creation of vacancies along with the presence of S on the surface enhanced the H2S adsorption energy slightly. However, addition of OH in the system caused H2S to bind on the surface sufficiently strongly. Additionally, H2S decomposition was found to be a spontaneous process in the presence of OH radicals.
1 Introduction
Hydrogen sulphide (H2S) is an essential factor of tail gas poising in many industrial processes and is one of the major products extracted during fuel gas processing.1,2 The removal of H2S from the tail gas and many industrial emissions using coal and sulphur products has gained a lot of importance recently because of its toxicity and adverse effects. Due to growing awareness about the depleting environmental resources, the environmental protection agencies are imposing more and more stringent air pollution regulations which include release of gases containing sulphur as well.3 A lot of research is being carried out throughout the world making use of various semiconductors for degrading pollutants. Among them, TiO2 in the form of anatase has attracted wide interest due to its strong oxidizing power under ultra violet (UV) irradiation, its chemical stability and absence of toxicity.4–9 Additionally, due to its high activity in degrading organic and inorganic air pollutants, titanium dioxide is used as a photocatalyst. It is biologically and chemically inert and cheaper as well.10–12Canela and co-authors have carried out a study making use of the TiO2 nanoparticles for studying the gas phase destruction of H2S gas under UV and visible light conditions and found it to be quite efficient.13 In another study conducted by our group to investigate the catalytic potential of TiO2 nanoparticles, we also observed a significant H2S destruction efficiency at high temperatures.14,15 We also carried out studies to evaluate the photocatalytic potential of TiO2 nanofibers and found them to be considerably efficient for the destruction of H2S gas.16
Adsorption and dissociation of H2S has been extensively studied on various metals including Cu (111) and (100), where dissociation of H2S resulted when Cu attracted one of its H atom.17,18 H2S decomposition on ZnO surface concluded in H2O formation.19 Masoud and co-authors found up to seven molecules of H2S binding to Pt atom.20 Additionally, various transition metals were taken into consideration for H2S decomposition reported in ref. 21 and the process was found facile over them. Huang and co authors studied the adsorption and reaction of H2S on TiO2 rutile (110) and anatase (101) surfaces.22 The authors suggested two different pathways for H2S dissociation on TiO2 surface resulting in formation of H2O and H2, respectively, which was comparatively harder for rutile (110) surface than anatase (101) due to lower energy barrier in the latter case.
Our experimental studies and findings are based on TiO2 nanoparticles and nanofibers in anatase phase. We have carried out these studies because metastable anatase form is found to be stable at nanoscale,23,24 because of relatively low surface energies of its free (101) and (100) facets.22,25–27 Rutile phase is most stable for larger diameters.24 Although, (101) faces in anatase are dominantly present, (001) surface exposes potentially more reactive facet having surface energy of 0.98 J m−2 because of stress on surface cleavage. This energy is almost double the value of ∼0.5 J m−2, found for stable (101) facet.28 In addition, many theoretical studies found (001) surface to be more reactive.29,30 Moreover, for (001) surface, the two fold coordinated oxygen atoms (indicated as O2c in Fig. 1) with 156° Ti–O–Ti angles are prone to reaction with adsorbates.28
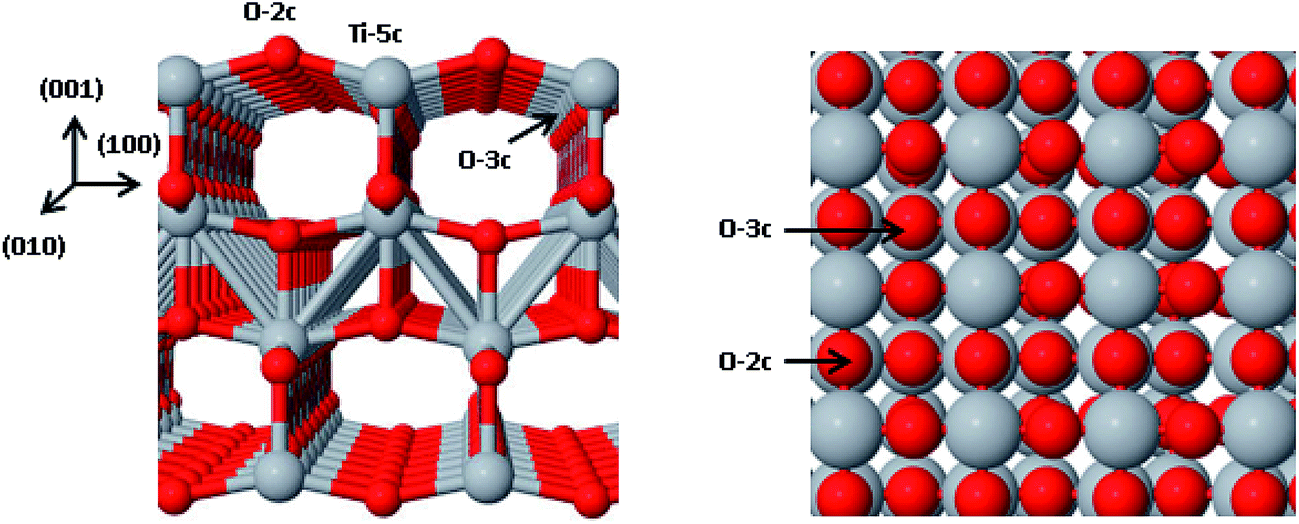 | ||
| Fig. 1 TiO2 slab and top view of (001) surface of anatase showing different positions of Ti and O atoms. | ||
After exploring the H2S destruction potential of pure and doped TiO2 nanocatalysts using various catalytic and photocatalytic experiments as narrated above, we used density functional theory (DFT) studies to further evaluate the H2S destruction and validate our experimental findings on the TiO2 anatase (001). The aim of the present work is to explore the adsorption and dissociation mechanism of H2S. The effect of co-adsorption of various species and defect creation has been evaluated on adsorption energy and destruction of H2S.
2 Computational details
Our calculations were carried out using the Vienna ab-initio simulation package (VASP)31 which uses plane wave as basis set to perform an iterative solution of the Kohn–Sham equations. Plane waves with a kinetic energy ≤400 eV were included in the calculations. The generalized gradient approximation (GGA) of Perdew and Wang (PW91) formalism was used to calculate the exchange–correlation energy.32 Blöchl's projector-augmented wave (PAW) method was used to describe the electron–ion interactions for Ti, O, S and H atoms.33 This is essentially a scheme combining the accuracy of all electron methods and the computational simplicity of the pseudo potential approach.34P(2 × 2) unit cell of anatase (001) which contains four Ti5c atoms on the surface, has been used in these computations. Each of these Ti5c atom is bonded to two raised 2c and two 3c lowered oxygen atoms in the (100) and (010) directions, respectively. Unit cell consists of 16 TiO2 units and periodically repeated slabs which are separated by ∼12 Å width. The reciprocal space for the p(2 × 2) unit cell was sampled with (4 × 4 × 1) k-point meshes generated automatically using the Monkhorst–Pack method.35 The upper half of the Ti and O atoms in the unit cell were relaxed. The relative positions of the Ti ions were fixed initially as those in the bulk, with optimized lattice parameters of 3.80 Å and 9.49 Å (the experimental values are 3.78 Å and 9.51 Å).36 The optimized lattice parameter was calculated using the tetragonal anatase unit cell and its reciprocal space was sampled with a (8 × 8 × 3) k-point grid. A first-order Methfessel–Paxton smearing-function with a width ≤0.1 eV was used to account for fractional occupancies.37
Partial geometry optimizations were performed using the RMM-DIIS algorithm38 and were stopped when all the forces were <0.05 eV Å−1. Vibrational frequencies for transition states (TS) were calculated within the harmonic approximation. Hessian matrix of the adsorbate was calculated neglecting the adsorbate-surface coupling.39 The climbing image nudged elastic band (cNEB) method was used in this study to determine minimum energy paths.40 Closed shell H2S and H2O molecules were optimized at the gamma point by non-spin polarized calculations using a 10 × 10 × 10 Å3 cubic unit cell. Spin-polarized calculations in a 10 × 12 × 14 Å3 orthorhombic unit cell were used for open shell species, H, O, S, OH and HS. All the calculations were performed at T = 0 K.
The slab of TiO2 (001) is shown in Fig. 1. The face (001) consists of periodically repeated slabs consisting of 16 units. The bond angles between Ti5c–O2c–Ti5c, Ti5c–O3c–Ti5c are 150 Å and 156.9 Å, respectively, while the distances between Ti5c–O2c, Ti5c–O3c are 1.97 Å and 1.94 Å, respectively.
The computations to determine the most stable adsorption configuration for H2S, HS, S, H and OH were carried out by placing each of them on four different possible locations, namely, hollow, five-fold bridging titanium (Ti5c), two-fold bridging oxygen (O2c) and three-fold bridging oxygen (O3c). The adsorption energies (Eads) were calculated by using the following equation:28
| Eads = [Ea/s − Es − nEa(gas)]/n | (1) |
3 Results and discussions
3.1 Adsorption of intermediates and H2S on the perfect surface
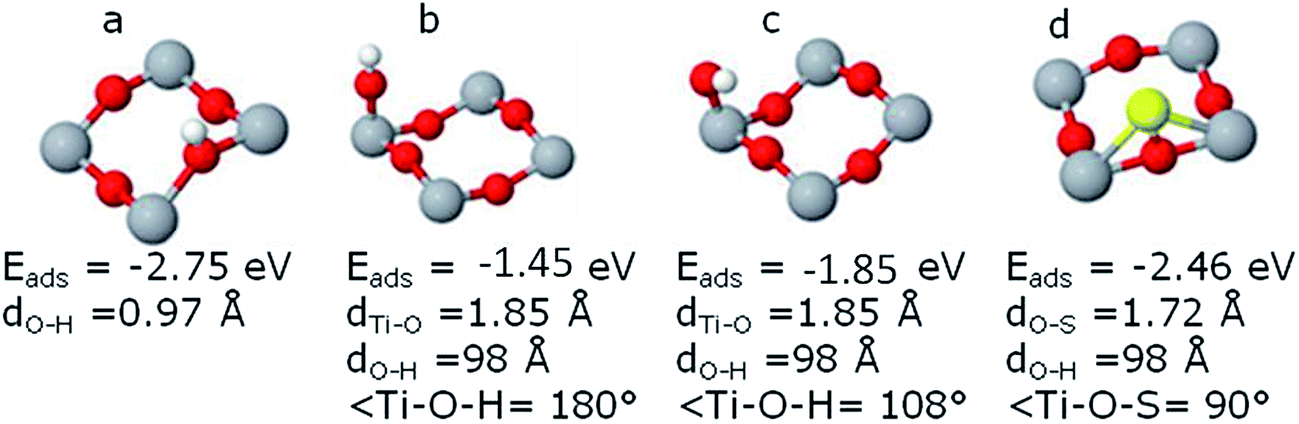 | ||
| Fig. 2 Adsorption properties of (a) H-atom, (b) OH, (c) OH (tilted) and (d) S-atom. The cases having maximum adsorption energies are presented in the figure only. | ||
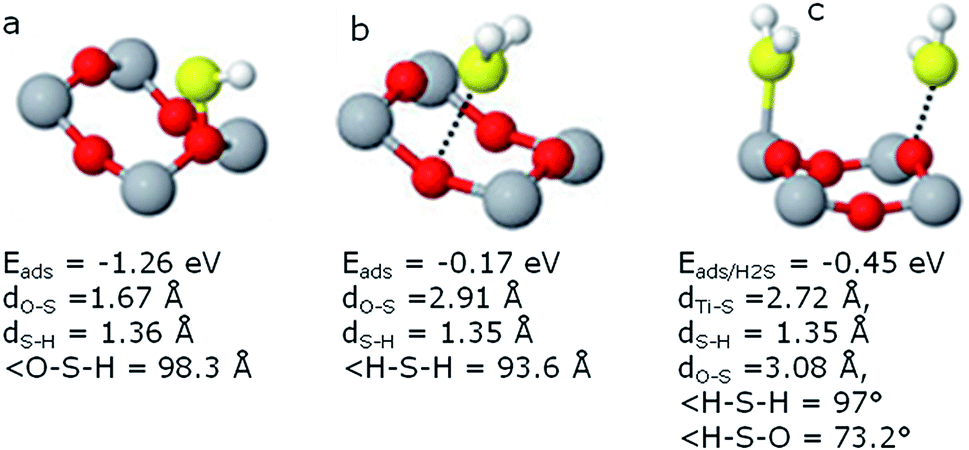 | ||
| Fig. 3 Most stable geometry configurations of (a) HS (tilted), (b) H2S and (c) H2S–H2S on TiO2 surface along with their adsorption energies. | ||
Additionally, the coverage effect on the adsorption energy of H2S has also been computed by placing two H2S molecules in one unit cell. H2S–H2S co-adsorption was investigated keeping both H2S on adjacent Ti5c positions as well as one H2S on Ti5c and the other on O2c. The adsorption energy per H2S molecule increased significantly, as compared to the energies obtained by placing single H2S molecule in the unit cell. Eads increased from −0.13 to −0.25 eV per molecule when both H2S were placed on adjacent Ti5c locations, while from +0.04 to −0.45 eV for second configuration showing significant enhancement due to coverage increase. The dipole–dipole interaction between the polar H2S molecules might be responsible for this increased stability. The hydrogen bonding might also be involved in stabilizing the system on doubling coverage. As shown in Fig. 3(c), the H2S molecule on O2c site is tilted bound making an angle H–S–O of 73.2°. The Ti5c–S distance is 2.72 Å and the H–S distance is 1.35 Å having an angle H–S–H at 97°. The S–O2c distance is 3.08 Å. The adsorption energies, bond lengths and angles measurements have been tabulated in Table 1. It can be inferred that H2S molecules have attractive interaction and the system stabilizes substantially for the second configuration.
| Surface | Species | Site | Eads |
|---|---|---|---|
| H | O2c | −2.75 | |
| OH | Ti5c (tilted) | −1.85 | |
| S | O2c | −2.46 | |
| HS | Hollow | −1.26 | |
| H2S | O3c | −0.17 | |
| H2S–H2S (per molecule) | Ti5c–O2c | −0.47 | |
| S–H2S | O2c–O3c | −0.91 | |
| S–HS | O2c–O3c | −0.9 | |
| H2S + OH | No S, H2S–O2c, OH–Ti5c | −0.33 | |
| H2S + OH-90 | No S, H2S–Ti5c, with H2S 90° rotated, OH–O2c | −0.09 | |
| S + H2S + OH | S–O2c, H2S–O2c, OH–Ti5c | −1.54 | |
| S + H2S + OH-90 | S–O2c, H2S–O2c, with H2S 90° rotated, OH–O2c | −1.07 | |
| Defected surface | H2S | No S, H2S–Ti5c | −0.20 |
| H2S | No S, H2S–O2c(vac) | −0.27 | |
| H2S | No S, H2S–O3c | −0.31 | |
| S + H2S | S–O2c, H2S–O2c(vac) | −0.21 | |
| S + H2S | S–O2c, H2S–Ti5c | −0.24 | |
| S + H2S | S–O2c, H2S–O3c | −0.96 | |
| S + H2S + OH | S–O2c, H2S–Ti5c, OH–O2c | −4.64 | |
| S + H2S (90°) + OH | S–O2c, H2S–O2c(vac), with H2S 90° rotated, OH–O2c | −2.67 | |
| H2S (90°) + OH | No S, H2S–O2c(vac), with H2S 90° rotated, OH–O2c | −2.25 | |
| Perfect surface TiO2 (p2 × 2) | S + HS + H(l) | S–O2c, one H of H2S–Ti5c on left O2c | −1.49 |
| S + HS + H(r) | S–O2c, one H of H2S–Ti5c on right O2c | −1.49 | |
| S + S + H + H | S–O2c, one H each of H2S–O3c on left O2c and on right O2c | ||
| Defected surface | S + HS + H(lt) | S–O2c, one H of H2S–Ti5c on left O2c(vac) | −1.6 |
| S + HS + H(rt) | S–O2c, one H of H2S–Ti5c on right O2c | −1.6 | |
| HS (90°) + H2O | No S, H2S–O2c(vac), with H2S 90° rotated and one H of H2S added to OH–O2c | −3.62 | |
| S + HS (90°) + H2O | S–O2c, H2S– O2c(vac), with H2S 90° rotated one H of H2S added to OH–O2c | −3.79 |
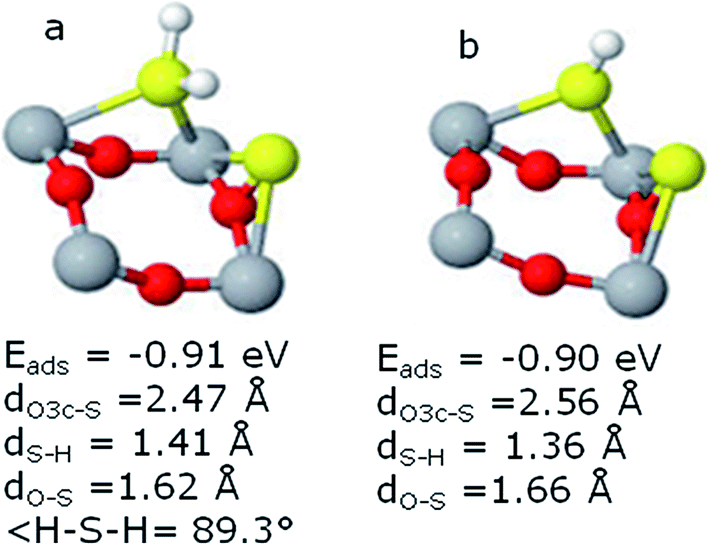 | ||
| Fig. 4 Adsorption properties of H2S and HS systems in the co-adsorbed state with S-atom are given in part (a) and (b), respectively. | ||
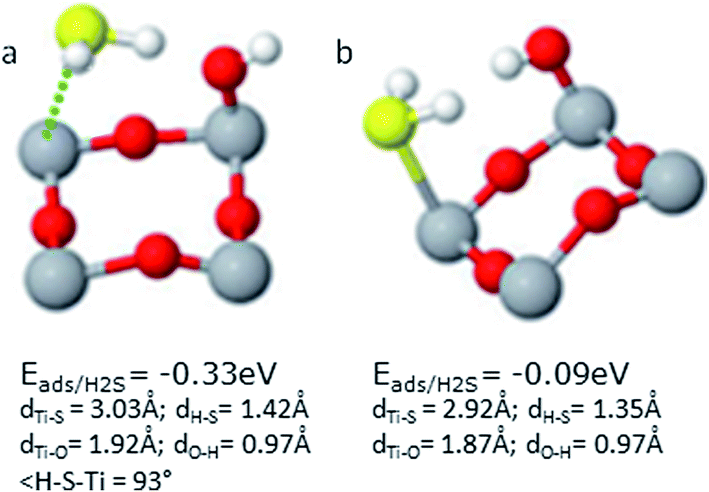 | ||
| Fig. 5 Adsorption properties of H2S in presence of OH. In (a) H of H2S points towards O-atom of OH. In (b) H2S is rotated at 90° and OH stands rotated at 180° relative to its position indicated in part (a). | ||
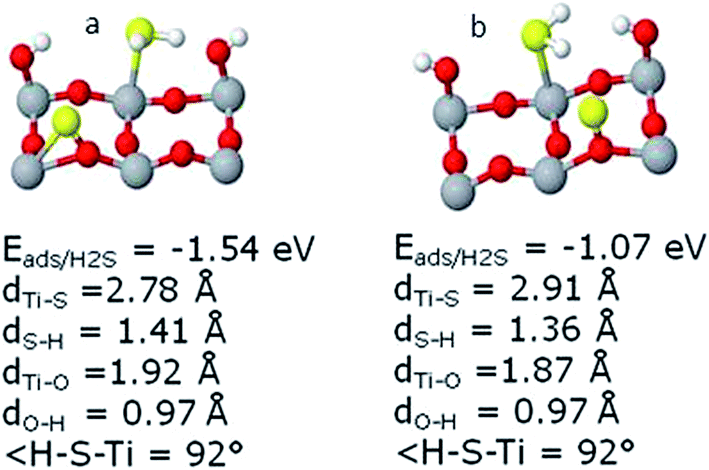 | ||
| Fig. 6 Adsorption geometries of H2S in presence of OH and S. In (b) part H2S is rotated at 90° relative to its orientation in part (a) in addition to change in position of S and OH direction where O–H stands rotated at 180°. | ||
In the second case where H2S was rotated at 90° as shown in Fig. 6(b), we observe a considerable difference in geometries if we compare to the same system in the absence of S as discussed previously. Both of H atoms in H2S and OH are tilted towards each other. The S–Ti5 distance is 2.91 Å and in this case the H atoms of H2S became almost parallel to the surface having angles H–S–Ti5c of 91.76° for both the H atoms, while H2S is not tilted at all. The H–S–H angle is 91.4° and H–S distance of 1.36 Å for both H atoms. The distance O–Ti5c and O–H is 1.87 and 0.97 Å, respectively. The O–H–Ti5c angle is measured to be 113.2°. S-atom is not tilted in this case and S–O2c distance is 1.63 Å.
It can be seen that in this system, the rotation of H2S by 90° had a negative effect on the H2S adsorption energies as it decreased from −0.33 to −0.09 eV without S and from −1.54 to −1.07 eV in the presence of S. It can be understood that H2S rotation is not favored in this system while the previously mentioned configuration is the most stable one. In addition, the lesser distance between S of H2S and the Ti5c in the former case can be attributed to a stronger adsorption. Finally, the most important is the OH orientation, which allows us to conclude that pointing of H of H2S towards O of OH plays key role in the system stability.
3.2 Defect (in TiO2 surface) creation effect on adsorption energies of various adsorbents
In order to explore the effect of vacancies on H2S adsorption or dissociation, a defect was created in the lattice of (2 × 2) TiO2 unit cell by removing a two way coordinated O2c atom, as shown in Fig. 7(a). Different calculations were carried out for various systems as discussed below.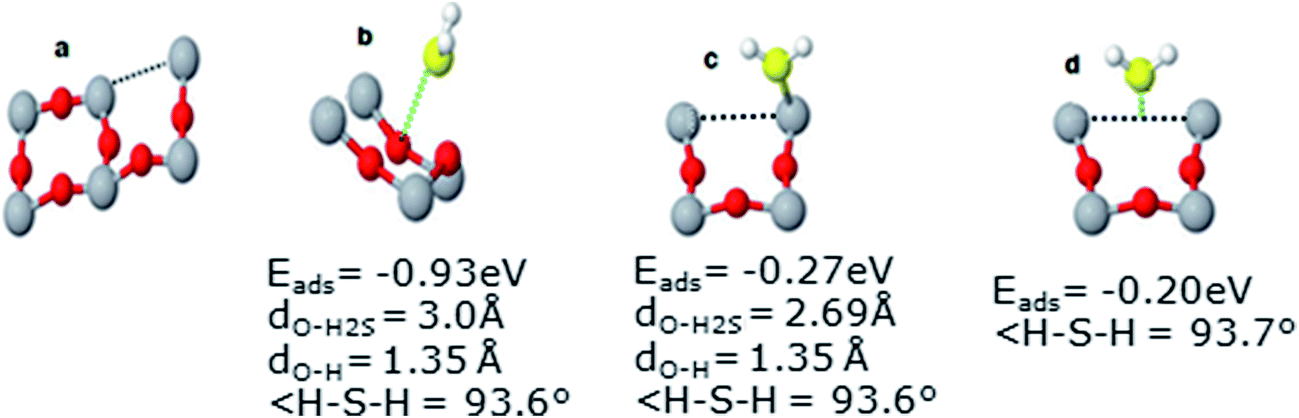 | ||
| Fig. 7 Different geometries of H2S adsorption in the presence of vacancy; (a) slab, (b) H2S on O3c, (c) H2S on Ti5c and (d) H2S on O2c (vacancy site). | ||
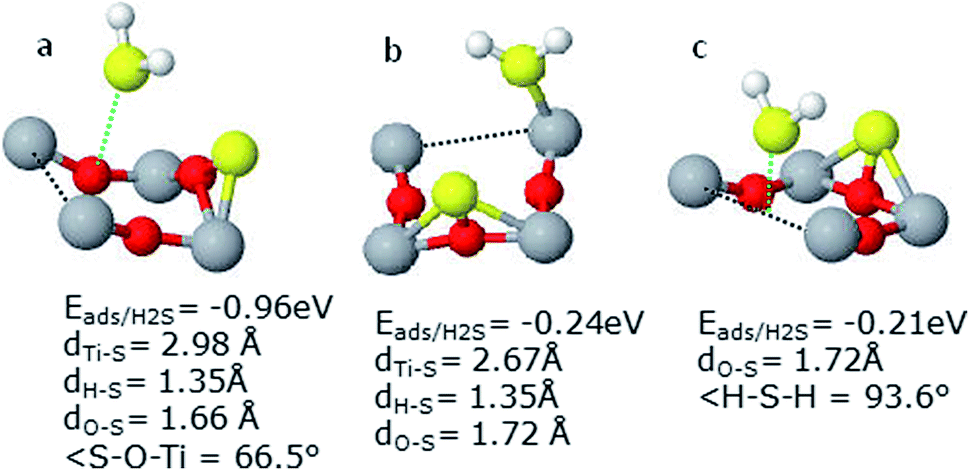 | ||
| Fig. 8 Different geometries in presence of vacancy and S, (a) H2S on O3c, (b) H2S on Ti5c and (c) H2S on O2c (vacancy site). | ||
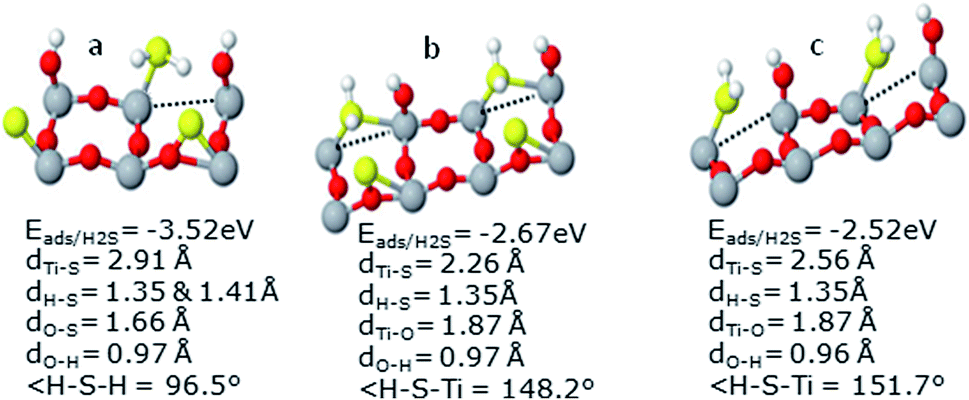 | ||
| Fig. 9 Adsorption properties of H2S on defected surface in the presence of S and OH depicted in part (a) and (b). In (b) H2S stands rotated at 90° relative to (a), while in (c) S-atom has been removed. | ||
Moreover, substantial stability of the system on OH addition in the presence of vacancy agrees with our previous findings on perfect surface. Here the H-atom gets tilted towards OH as it was noted in the case of perfect surface. To evaluate the actual cause of increase in the H2S adsorption energy, further computations were carried by removing OH from the system, as depicted in Fig. 9(b). It was found that the H2S adsorption energy decreased to almost fifty percent i.e., −1.31 eV as compared to −2.67 eV when OH was present in this system. It can therefore be deduced from these findings that OH addition has a substantial effect towards the adsorption energy of H2S. The effect becomes quite pronounced in the presence of vacancy. In agreement with our findings on perfect surface, orientation of the molecule and OH is of crucial importance.
Just to further ascertain these findings, calculations were also carried out without S-atom. H2S molecule rotated at 90° relative to the system given in Fig. 9(a) (as shown in Fig. 9(c)) yielded lower Eads of H2S (decreased from −2.67 to −2.25 eV). However, this decrease is in line as observed on the perfect surface. In addition, it highlights the significant rise in the Eads of H2S which was caused by addition of OH. This very high Eads of H2S may cause poisoning of the catalytic surface. Hence, OH radicals must be avoided on the defected surface to save the catalyst from H2S poisoning. Detailed geometries have been shown in Fig. 9.
3.3 Dissociation of H2S
Looking at the affinity of OH towards H-atom of H2S as found in these computations, it seems to be more likely that when H2S will dissociate in the presence of OH, one H-atom of H2S will join OH to form water. The investigations of this hypothesis were carried out on perfect and defected surface by placing HS with OH in the system in the presence of S.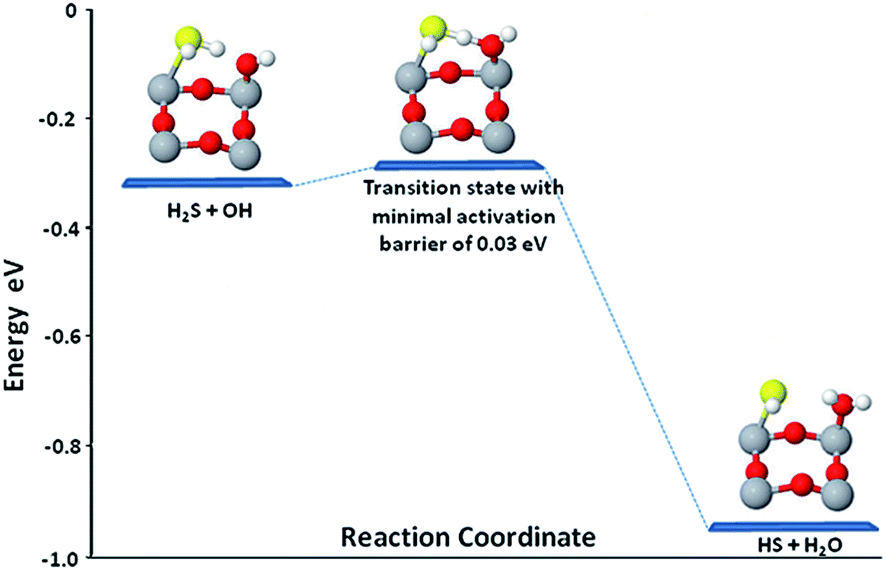 | ||
| Fig. 10 Energy profile for the activation of H2S on anatase (001). | ||
Additionally HS fragment of H2S moved to stay on Ti5c with HS (H atom which has been detached from H2S) distance of 1.89 Å. The resulting rotated HS is tilted making an angle of H–S–Ti5c of 94.2°. The decomposition of H2S and formation of H2O also lead to some structural changes in the co-adsorbed S and associated O2c on the surface. The detailed adsorption energies along with all the geometries are presented in Table 1S (ESI†). Thus, in the presence of vacancy, H2S decomposition is a spontaneous process. The negligible activation barriers of 0.05 eV were found in our experimental observations.48 Thus, our DFT results are in complete agreement with the experimental findings for the S-doped nanofibers with highest H2S destruction efficiency.45
4 Conclusions
Interaction of H2S, its dissociated products and OH radical were studied on anatase phase of TiO2 (001) surface implying DFT calculations at GGA level of approximation. The aim of the present work is to optimize H2S adsorption and study its destruction on the surface inspired by the experimental observations carried out in our previous work.45 The perfect surface weakly interacts with H2S. However, the presence of S on the surface led to a strong H2S binding on a particular position O3c by giving an adsorption energy of −0.91 eV. Increase in H2S coverage also helped to stabilize the molecules to some extent. Defect creation in the surface further stabilized the molecule slightly. Nevertheless, the most significant impact was experienced due to the presence of OH radicals in the vicinity, which caused the adsorption energy of H2S to enhance for both the perfect and defected surfaces. S, HS, OH species interact sufficiently strongly with this surface. The presence of OH radicals on the surface help to decompose H2S into HS and H2O (H2S + OH → HS + H2O) instantaneously. Thus anatase (001) surface can readily destroy H2S species if moisture is present in the system. On the other hand, it was reported in a recent study that hydroxylated anatase (001) leads to Ti3+ formation from the Ti–O bond cleavage. This is beneficial for reactions of H2O and O2 in addition to its prime favorability for ethanol dissociation.41,49 Therefore, further studies should be carried out to explore the effects of Ti3+ for H2S adsorption as an extension to this study.Conflict of interest
No competing interests in the publication of this paper.Author's contribution
NS carried out the experimental studies and the computational studies as well. He was involved in preparing the draft and final manuscript. AH conceived the study, and participated in its basic understanding by the author to carry out this study. He also participated in helping the author to draft this manuscript and final vetting of the manuscript. NM, MBK and SGS helped the authors for the preparation of draft manuscript. All authors read and approved the final manuscript.Acknowledgements
Authors from Pakistan gratefully thank the National Centre for Physics (NCP), Islamabad, Pakistan, Nanoscience and Catalysis division and DNE, PIEAS, Islamabad labs and facilities for carrying out this study.References
- M. J. Pearson, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 154–158 CAS.
- Z. M. George, Phosphorus Sulfur Relat. Elem., 1976, 1, 315–322 CrossRef CAS.
- E. Laperdrix, G. Costentin, N. Nguyen, F. Studer and J. C. Lavalley, Catal. Today, 2000, 61, 149–155 CrossRef CAS.
- A. Fujishima, T. N. Rao and T. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- A. G. Agrios and P. Pichat, J. Appl. Electrochem., 2005, 35, 655–663 CrossRef CAS.
- M. Lewandowski and D. F. Ollis, in Seminoconductor photochemistry and photophysics, ed. V. Ramamurthy and K. S. Schanze, Marcel Dekker, New York, 2004, pp. 249–282 Search PubMed.
- P. Pichat, in Chemical degradation methods for wastes and pollutants: environmental and industrial applications, ed. M. A. Tarr, Marcel Dekker, New York, 2003, pp. 77–119 Search PubMed.
- H. Ibrahim and H. Lasa, Appl. Catal., B, 2002, 38, 201–213 CrossRef CAS.
- J. Zhang, A. Terukazu, M. Minagawa, K. Kinugawa, H. Yamashita, M. Matsuoka and M. Anpo, J. Catal., 2001, 198, 1–8 CrossRef CAS.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–8 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–743 CrossRef CAS.
- A. Fujishima and X. Zhang, C. R. Chimie, 2006, 9, 750–760 CrossRef CAS.
- M. C. Canela, M. A. Rosana and W. F. Jardim, J. Photochem. Photobiol., A, 1998, 12, 73–80 CrossRef.
- N. Shahzad, S. T. Hussain, A. Siddiqua and M. A. Baig, J. Nanosci. Nanotechnol., 2012, 12(6), 5061–5065 CrossRef CAS.
- N. Shahzad, S. T. Hussain and A. Nisar, Chalcogenide Lett., 2013, 10(1), 19–26 CAS.
- N. Shahzad and C. Qiao, Mater. Sci. Forum, 2013, 756, 225–230 CrossRef.
- L. Min, Z. Jun-ying, Z. Yue, Z. Guo-feng and W. Tian-min, Appl. Surf. Sci., 2011, 257, 10710–10714 CrossRef.
- C. Shenghui, S. Shuangqing, L. Bingjie, M. Yunfei, Y. Youguo and H. Songqing, Surf. Sci., 2014, 620, 51–58 CrossRef.
- L. Lixia, Z. Riguang, H. Peide and W. Baojun, Fuel Process. Technol., 2013, 106, 222–230 CrossRef.
- D. G. Masoud, S. Narges, A. Mahdi and G. A. Morteza, Appl. Surf. Sci., 2012, 261, 697–704 CrossRef.
- T. V. Reshetenko, S. R. Khairulin, Z. R. Ismagilov and V. V. Kuznetsov, Int. J. Hydrogen Energy, 2002, 27, 387–394 CrossRef CAS.
- W. F. Huang, H. T. Chen and M. C. Lin, J. Phys. Chem. C, 2009, 113, 20411–20420 CAS.
- M. R. Ranade, A. Navrotsky, H. Z. Zhang, J. F. Banfield, S. H. Elder, A. Zaban, P. H. Borse, S. K. Kulkarni, G. S. Doran and H. J. Whitfield, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6476–6481 CrossRef CAS.
- A. S. Barnard and P. Zapol, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235403 CrossRef.
- A. S. Barnard, P. Zapol and L. Curtiss, Surf. Sci., 2005, 582, 173–188 CrossRef CAS.
- C. Arrouvel, M. Digne, M. Breysse, H. Toulhoat and P. Raybaud, J. Catal., 2004, 222, 152–166 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- H. Akhtar, PhD thesis, Eindhoven University of Technology, Netherlands, 2010.
- X. Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560–19562 CrossRef CAS PubMed.
- A. Vittadini, M. Casarin and A. Selloni, Theor. Chem. Acc., 2007, 117, 663 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(23), 13244–13249 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13(12), 5188–5192 CrossRef.
- M. Horn, C. F. Schwerdt and E. P. Meagher, Z. Kristallogr., 1972, 136(3–4), 273–281 CrossRef CAS.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40(6), 3616–3621 CrossRef CAS.
- P. Pulay, Chem. Phys. Lett., 1980, 73(2), 393–398 CrossRef CAS.
- J. D. Head, Int. J. Quantum Chem., 1997, 65(5), 827–838 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- Z. Riguang, L. Zhixue, L. Lixia and W. Baojun, Appl. Surf. Sci., 2015, 353, 150–157 CrossRef.
- F. Lin, G. Zhou, Z. Li, J. Li, J. Wu and W. Duan, Chem. Phys. Lett., 2009, 475, 82–85 CrossRef CAS.
- A. Hussain, J. Gracia, B. E. Nieuwenhuys and J. W. Niemantsverdriet, ChemPhysChem, 2010, 11(11), 2375–2382 CrossRef CAS PubMed.
- M. Menetrey, A. Markovits and C. Minot, Surf. Sci., 2003, 524(1–3), 49–62 CrossRef CAS.
- N. Shahzad, PhD thesis, National University of Sciences and Technology (NUST), Pakistan, 2010.
- M. E. Amani, J. Jacek and J. B. Teresa, J. Mater. Chem. A, 2015, 3, 8194–8204 Search PubMed.
- O. Mabayoje, M. Seredych and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2012, 4(6), 3316–3324 CAS.
- N. Shahzad and A. Nisar, Chalcogenide Lett., 2015, 12, 129–136 CAS.
- Y. Wang, H. J. Sun, S. J. Tan, H. Feng, Z. W. Cheng, J. Zhao, A. D. Zhao, B. Wang, Y. Luo, J. L. Yang and J. G. Hou, Nat. Commun., 2013, 4, 2214 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional data file is a table giving detailed adsorption energies along with all the geometries and presented as Table 1S. See DOI: 10.1039/c5ra20875k |
| ‡ NS is working as Assistant Professor at National University of Sciences and Technology, Islamabad, Pakistan. |
| This journal is © The Royal Society of Chemistry 2016 |