
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of functionalized molecular and macromolecular double-decker silsesquioxane systems†
P.
Żak
a,
M.
Majchrzak
a,
G.
Wilkowski
a,
B.
Dudziec
a,
M.
Dutkiewicz
b and
B.
Marciniec
*b
aDepartment of Organometallic Chemistry, Faculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89B, 61-614 Poznan, Poland
bCentre for Advanced Technologies, Adam Mickiewicz University in Poznan, Umultowska 89C, 61-614 Poznan, Poland. E-mail: Bogdan.Marciniec@amu.edu.pl
First published on 26th January 2016
Abstract
Synthesis and characterization of new, molecular DDSQ-based compounds functionalized by substituted styrenes (also with heteroatoms) in a stereoselective manner is presented. Additionally, a novel class of vinylene-arylene linear copolymers with double-decker silsesquioxane fragments in the main chain is developed. These molecular and macromolecular compounds were synthesized via efficient and highly stereoselective silylative coupling (and copolycondensation) and cross-metathesis (and ADMET) reactions.
Introduction
Polyhedral oligomeric silsesquioxanes (POSS) of the empirical formula (RSiO1.5)n with different reactive and/or inert organic coronae, have recently become the subject of growing interest in many branches of chemistry.1 It is known that cage structure silsesquioxanes, due to their unique properties, e.g. the rigid inorganic SiO core with a diameter of about 1.5 nm, are often regarded as the smallest silica particles that can also influence the surface properties of functional groups attached to silicon atoms. A well-defined organization of POSS is important because of their possible use as precursors and components of versatile inorganic/organic hybrid materials that display nanophase behavior. Not more than a decade ago, a new class of silsesquioxyl compounds containing rigid Si–O–Si bonds, i.e. the double-decker silsesquioxane (DDSQ), was reported by Yoshida and co-workers.2 The structure of DDSQ is an open cage that includes two cyclosiloxane rings in parallel planes with 8 inert phenyl groups at the silicon atoms of each ring. These two rings are joined by oxygen bridges. Current research on that compound refers to two major trends in its development and knowledge, i.e. syntheses of molecular and macromolecular organosilicon compounds based on DDSQ that may exhibit interesting and desired physicochemical properties.3The silylative coupling (SC) and cross-metathesis (CM) of olefins with vinyl-substituted organosilicon compounds have been studied and developed for the last two decades as a new, effective catalytic activation of the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H bond of olefins and the C–Si bond of organosilicon compounds (Scheme 1). The mechanisms of these reactions are diverse as they require different catalysts (mostly ruthenium hydride for SC and ruthenium-carbene for CM). However, both SC and CM proved to be effective paths for synthesis of functional vinylsilicon molecular as well as macromolecular compounds.4
C–H bond of olefins and the C–Si bond of organosilicon compounds (Scheme 1). The mechanisms of these reactions are diverse as they require different catalysts (mostly ruthenium hydride for SC and ruthenium-carbene for CM). However, both SC and CM proved to be effective paths for synthesis of functional vinylsilicon molecular as well as macromolecular compounds.4
 | ||
| Scheme 1 Difference in regioselectivity of silylative coupling (SC) and cross-metathesis (CM) reactions of vinylsilanes with olefins. | ||
In the previous decade, we reported both processes (SC and CM) to be complementary routes for regio- and stereoselective synthesis of functionalized vinylsilsesquioxanes and vinylspherosilicates.5 Cross-metathesis of vinylsilsesquioxanes using Grubbs catalyst was exploited by Feher,6 Laine,7 Cole-Hamilton and co-workers8 as well as Naka.9 Schrock noted the use of molybdenum catalyst for CM but in all reactions, except for styrene, a mixture of stereoisomers was obtained.6 On the other hand, hydrosilylation and Heck reaction were applied for vinyl-substituted POSS modification to obtain compounds of interesting photophysical properties enabling their use as sugars and peptide scaffolds.8a,10
Our recent report has proved a successful use of divinyl-substituted double-decker silsesquioxanes (DDSQ-2SiVi1A and 1B) as substrates for highly effective silylative coupling (SC) and/or cross-metathesis (CM) with olefins (Scheme 2).11 The reactions led to highly stereoselective formation of E isomers. Preliminary studies on silylative coupling copolycondensation (SCC) of divinylbenzene (DVB) with DDSQ-2SiVi show a possibility to obtain stereoregular, linear cooligomer containing DDSQ-vinylene-phenylene units in the chain.
 | ||
| Scheme 2 Protocol using DDSQ-2SiVi for stereoselective silylative coupling (also copolycondensation) and cross-metathesis with olefins. | ||
Here, we present a synthetic path for obtaining new, molecular DDSQ-based compounds functionalized by substituted styrenes (also with heretoatoms) in a stereoselective manner as well as a new class of vinylene-arylene copolymers with double-decker silsesquioxane in the main chain. All of the compounds were characterized via spectroscopic methods and their structural properties were also studied.
Experimental
Materials and methods
All reactions were carried out under argon atmosphere using standard Schlenk-line and vacuum techniques. All solvents were dried prior to use over CaH2 and stored under argon over molecular sieves type 4 Å. Dichloromethane was additionally passed through a column with alumina and after that it was degassed by repeated freeze–pump–thaw cycles.The chemicals were purchased from the following sources: dichloromethane (DCM), tetrahydrofuran (THF), n-hexane, chloroform-d, dichloromethane-d2, ethanol, methanol, acetone, calcium hydride, diethyl ether, tricyclohexylphosphine, 9-bromoanthracene, 1-bromonaphthalene, 2-bromothiophene, 2,5-dibromothiophene, bromopentafluorobenzen, 4-vinylphenylboronic acid, 1,4-dibromo-2,3,5,6-tetrafluorobenzene, 9,10-dibromoanthracene, 1,4-dibromonaphthalene, molecular sieves type 4 Å, copper(I) chloride and anhydrous magnesium sulfate from Aldrich, vinylmethyldichlorosilane and vinylphenyl-dichlorosilane from ABCR, Grubbs first generation benzylidene catalyst from Apeiron Synthesis, triethylamine and silica gel 60 from Fluka, ruthenium(III) chloride hydrate from Lancaster, tetrasilanolphenyl POSS (Ph8O8Si8)(OH)4(DDSQ-4OH) from Hybrid Plastics. Divinyl-substituted double-decker silsesquioxanes (DDSQ-2SiVi) (1A and 1B) were prepared according to the literature procedure.11b All mono- and distyrylarenes were synthesized according to the literature procedure.12 The ruthenium species [RuHCl(CO)(PCy3)2] was made in accordance with the procedure described in literature.13
Techniques and measurement
General procedure for the synthesis of functionalized E,E-vinylene-arylenesilsesquioxanes (1–8)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 1:5) to remove ruthenium complexes. Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
:CH2Cl2 = 1:5) to remove ruthenium complexes. Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
System 1. A 5 mL two-necked glass reactor equipped with a reflux condenser and connected to a bubbler was charged under argon with divinylsilsesquioxane (1A) (0.05 g, 4.13 × 10−5 mol), toluene (1 mL) and styrylarylene derivative (1.24 × 10−4 mol). The reaction mixture was heated up to 90 °C (or 110 °C for thiophene derivatives) and [RuHCl(CO)(PCy3)2] (0.0006 g, 8.26 × 10−7 mol) was added and the catalytic system was left for 24 h (except thiophene derivative for 96 h). Then the solvent was evaporated under vacuum and the crude product was dissolved in DCM (1 mL). After that, the brown solution was filtered at ‘flash-system’ (glass filter–SiO2–Celite) using n-hexane three times (3 × 10 mL) to remove the excess of olefin and the final product was washed by DCM (2 × 10 mL). Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
System 2. A 5 mL two-necked glass reactor equipped with a reflux condenser and connected to a bubbler was charged under argon with divinylsilsesquioxane (1A) (0.05 g, 4.13 × 10−5 mol), dichloromethane (2 mL) and styrylarylene derivative (8.26 × 10−5 mol). The reaction mixture was stirred and heated in an oil bath to maintain a gentle reflux (ca. 45 °C). Then the hydride ruthenium complex [RuHCl(CO)(PCy3)2] (0.0006 g, 8.26 × 10−7 mol) was added to the mixture under argon. After 5 min copper(I) chloride (0.0004 g, 4.13 × 10−6 mol) was added. The reaction mixture was heated under reflux for 24 h. Then the solvent was evaporated under vacuum and cold n-hexane (2 mL) was added to the remaining content to form precipitate. The precipitate was filtered off and purified by column chromatography (silica gel 60/n-hexane
:CH2Cl2 = 1:5) to remove ruthenium complexes. Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
Analytical data for isolated molecular compounds (1–8)
CH–Si), 7.05–8.61 (m, 78H, CH–C6H4– and aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 123.36, 125.19, 125.45, 126.62, 126.97, 126.83, 127.56, 127.60, 127.65, 127.71, 127.77, 127.94, 127.99, 128.47, 129.61, 130.44 (d, J = 4.4 Hz), 130.71, 131.51, 134.00, 134.05, 134.11, 134.16, 137.38, 140.12, 147.53, 165.39; 29Si NMR (79 MHz, CD2Cl2, δ (ppm)): −44.36, −77.75, −79.21.
CH–Si and CH–C6H4–), 7.02–8.28 (m, 72H, aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 122.31, 125.40, 125.82, 126.07, 126.84 (br s), 127.54, 127.61, 127.67, 127.70, 127.95, 128.25, 130.19, 130.34, 130.46 (br s), 130.65, 131.44, 131.56, 133.85, 133.99, 134.02, 134.06, 134.08, 134.15, 134.50, 136.42, 139.75, 141.11; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.37, −77.83, −79.31.
CH–Si), 6.97–8.15 (m, 60H, CH–C6F4 and aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 121.80, 124.13, 127.07, 127.59 (t, J = 5.9 Hz), 127.88, 127.95, 128.02, 130.26 (t, J = 4.9 Hz), 130.36, 130.41, 130.47, 130.67 (t, J = 2.4 Hz), 131.43 (t, J = 2.1 Hz), 133.94, 133.98, 134.02, 138.32, 147.08; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.78, −77.83, −79.33.
CH–Si), 7.05–8.28 (m, 66H, CH–C6H4, C6H5, C6H4 and C4H3S); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 122.15, 123.32, 125.12, 125.75, 127.44, 127.50, 127.59, 127.67, 127.93, 128.15, 129.60, 130.33 (t, J = 4.3 Hz), 130.44 (t, J = 3.0 Hz), 130.63, 131.52, 134.00 (d, J = 6.1 Hz), 134.47, 134.51, 136.56, 143.85; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.39, −77.89, −79.34.
CH–Si), 6.95–8.08 (m, 68H, CH–C6H4– and aromatic parts); 13C NMR (75 MHz, CDCl3, δ, ppm): −0.75, −0.65, 126.13, 126.24, 127.17, 127.30, 127.66, 127.74, 127.80, 128.01, 130.36, 130.49, 130.63, 130.88, 131.11, 132.00, 134.11, 134.27, 138.60, 145.63; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.91, −78.18, −79.28, −79.46, −79.52.
CH–Si), 6.88–7.94 (m, 64H, CH–C6H4– and aromatic parts); 13C NMR (75 MHz, CDCl3, δ, ppm): −0.69, −0.68, 125.45, 125.88, 125.99, 126.09, 126.80, 127.57, 127.64, 127.69, 127.79, 127.91, 128.39, 130.20, 130.36, 130.41, 130.47, 130.92, 131.57, 132.05, 133.86, 134.06, 134.26, 136.62, 139.93, 140.89, 146.29; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.13, −78.23, −79.07, −79.47, −79.86.
CH–Si), 6.86–7.61 (m, 50H, CH–C6F4– and aromatic parts); 13C NMR (75 MHz, CDCl3, δ, ppm): −0.84, −0.71, 126.13, 126.24, 127.17, 127.30, 127.66, 127.74, 127.93, 128.01, 130.37, 130.49, 130.57, 130.64, 130.88, 131.11, 132.01, 134.11, 134.27, 138.59, 145.61; 29Si NMR (79 MHz, CDC3, δ, ppm): −30.57, −78.26, −79.18, −79.53, −79.84.
CH–Si), 6.98–7.66 (m, 42H, aromatic parts, 2H coming from CH–C6H4– and 2H coming from –C4H3S–), 7.21 (d, 2H, JHH = 6.0 Hz, –C4H3S–), 7.53 (d, 4H, JHH = 7.2 Hz, –C6H4–), 7.70 (d, 2H, JHH = 9.0 Hz, –C4H3S–), 7.74 (d, 4H, JHH = 9.0 Hz, –C6H4–), 7.87 (d, 4H, JHH = 7.2 Hz, –C6H4–).; 13C NMR (75 MHz, CDCl3, δ, ppm): −0.62, −0.58, 123.30, 124.22, 125.12, 125.98, 127.47, 127.74, 127.97, 128.24, 130.55, 130.97, 132.10, 134.13, 134.27, 134.46, 136.87, 144.26, 146.07; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.17, −78.25, −79.20, −79.50, −79.77.
General procedure for the synthesis of functionalized E,E-arylene-silsesquioxanes derivatives oligomers (9–15)
:CH2Cl2 = 1:5) to remove ruthenium complexes. Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
System 1. A mixture of silicon monomer–divinylsilsesquioxane (1A) (0.05 g, 4.13 × 10−5 mol), distyrylarene (4.13 × 10−5 mol), [RuHCl(CO)(PCy3)2] (0.0006 g, 8.26 × 10−7 mol) and toluene (1.5 mL) was placed in a 5 mL two-necked glass reactor equipped with a magnetic stirring bar and condenser connected to a bubbler. The reaction mixture was heated at 110 °C and stirred for 72 h. The reaction progress was monitored by 1H NMR. After the coupling process was complete, the solvent was evaporated under vacuum and dichloromethane was added. Then, the final product was purified by repeated precipitation from methanol. The polymeric material was filtered off and dried under vacuum to give an analytically pure sample (white or yellow powder).
System 2. A mixture of silicon monomer–divinylsilsesquioxane (1A) (0.05 g, 4.13 × 10−5 mol), distyrylarene (4.13 × 10−5 mol), and dichloromethane (2 mL) was placed in a 5 mL two-necked glass reactor equipped with a magnetic stirring bar and condenser connected to a bubbler. The reaction mixture was heated up to 45 °C and [RuHCl(CO)(PCy3)2] (0.0006 g, 8.26 × 10−7 mol) was added. After 5 min copper(I) chloride (0.0004 g, 4.13 × 10−6 mol) was added and the reaction mixture was heated under reflux for 3 days. The reaction progress was monitored by 1H NMR. After the coupling process was complete, the solvent was evaporated under vacuum and cold n-hexane (2 mL) was added to the remaining content to form precipitate. The precipitate was filtered off and purified by column chromatography (silica gel 60/n-hexane
:CH2Cl2 = 1:5) to remove ruthenium complexes. Evaporation of the solvent gave an analytically pure sample (white or yellow powder).
Analytical data for isolated macromolecular compounds (9–15)
CH2 terminal), 6.62 (d, 1H, JHH = 18.3 Hz, CH–Si), 6.99–7.97 (m, 56H, aromatic parts and CH–C6H4–); 13C NMR (75 MHz, CDCl3, δ, ppm): 0.11, 0.14, 124.45, 126.04, 126.11, 126.31, 126.95, 127.45, 128.01, 130.36, 130.48, 132.02, 132.14, 134.16, 134.35, 136.81, 138.05, 139.74, 146.32; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.19, −78.27, −79.51.
CH–Si), 6.98–8.27 (m, 65H, CH–C6H4–, C6H5 and naphthalene); 13C NMR (125 MHz, CDCl3, δ, ppm): 122.71; 125.98, 126.36, 126.95, 127.46, 127.55, 127.63, 127.68, 127.75, 127.91 (br s), 129.76, 130.23, 130.32, 130.52, 130.65, 130.81, 131.74, 131.86, 134.10, 134.18, 134.23, 136.55, 139.55, 141.07, 147.71, 165.46; 29Si NMR (79 MHz, CDCl3, δ, ppm): −44.55, −77.90, −79.35.
CH2 terminal), 6.62 (d, 1H, JHH = 19.5 Hz, CH–Si), 6.91–7.81 (m, 58H, aromatic part and CH–C6H4–); 13C NMR (75 MHz, CDCl3, δ, ppm): −0.71, 125.25, 126.89, 127.07, 129.95, 130.61, 131.61, 134.17, 134.33, 136.98, 147.90; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.46, −78.27, −78.36, −79.51.
CH–Si), 7.03–8.74 (m, 68H, CH–C6H4– and aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 123.36, 125.15, 126.83 (t, J = 6.1 Hz), 127.56, 127.60, 127.65, 127.71, 127.77, 127.94, 127.99, 128.47, 129.61, 130.42, 130.47, 130.71, 131.51, 133.19, 134.00, 134.05, 134.11, 134.16, 137.38, 140.12, 147.53, 165.38; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.63, −77.77, −79.25.
![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C–HCCH2), 5.84 (d, 1H, JHH = 17.1 Hz, trace amount of signals coming from C–HCCH2), 6.55 (d, 1H, JHH = 19.2 Hz, CH–Si), 6.79 (dd, 1H, trace amount of signals coming from C–HCCH2), 6.88–7.76 (m, 49H, aromatic parts and 1H coming from CH–C6H4–); 13C NMR (75 MHz, CDCl3, δ, ppm): −1.13, −0.87, 125.95, 126.55, 127.11, 127.41, 127.76, 127.93, 128.01, 130.43, 130.51, 130.88, 132.04, 134.12, 134.26, 145.75; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.50, −78.20, −78.24, −78.95, −79.50.
CH–Si), 6.98–8.27 (m, 67H, CH–C6H4– and aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 123.93, 127.02, 127.61 (t, J = 8.6 Hz), 127.96, 130.30, 130.34, 130.37, 130.41, 130.49, 130.56, 130.67 (t, J = 3.7 Hz), 131.46, 134.00 (d, J = 7.9 Hz), 138.19, 147.21; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.71, −77.81, −79.31.
CH2 terminal), 6.46 (d, 1H, JH–H = 19.2 Hz, CH–Si), 6.85–7.80 (m, 52H, aromatic part and –Si–HCCH–); 13C NMR (125 MHz, CDCl3, δ, ppm): −1.14, −0.70, 124.21, 125.32, 125.53, 127.41, 127.62, 127.85, 128.24, 129.05, 130.42, 131.96, 134.01, 134.12, 134.22, 137.89, 145.89; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.15, −78.26, −79.41, −79.51.
C–HCCH2), 5.84 (d, 1H, JHH = 17.1 Hz, trace amount of signals coming from C–HCCH2), 6.55 (d, 1H, JHH = 19.2 Hz, CH–Si), 6.79 (dd, 1H, trace amount of signals coming from C–HCCH2), 6.88–7.76 (m, 49H, aromatic parts and 1H coming from CH–C6H4–); 13C NMR (75 MHz, CDCl3, δ, ppm): −1.13, −0.87, 125.95, 126.55, 127.11, 127.41, 127.76, 127.93, 128.01, 130.43, 130.51, 130.88, 132.04, 134.12, 134.26, 145.75; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.50, −78.20, −78.24, −78.95, −79.50.
CH–Si), 6.98–8.27 (m, 67H, CH–C6H4– and aromatic parts); 13C NMR (125 MHz, CD2Cl2, δ, ppm): 123.93, 127.02, 127.61 (t, J = 8.6 Hz), 127.96, 130.30, 130.34, 130.37, 130.41, 130.49, 130.56, 130.67 (t, J = 3.7 Hz), 131.46, 134.00 (d, J = 7.9 Hz), 138.19, 147.21; 29Si NMR (79 MHz, CD2Cl2, δ, ppm): −44.71, −77.81, −79.31.
CH2 terminal), 6.46 (d, 1H, JH–H = 19.2 Hz, CH–Si), 6.85–7.80 (m, 52H, aromatic part and –Si–HCCH–); 13C NMR (125 MHz, CDCl3, δ, ppm): −1.14, −0.70, 124.21, 125.32, 125.53, 127.41, 127.62, 127.85, 128.24, 129.05, 130.42, 131.96, 134.01, 134.12, 134.22, 137.89, 145.89; 29Si NMR (79 MHz, CDCl3, δ, ppm): −30.15, −78.26, −79.41, −79.51.
Results and discussion
Taking into regard to the positive results presented in our recent paper on the possibility of using cross-metathesis reaction and silylative coupling in functionalization of divinyl-substituted double-decker silsesquioxanes, we decided to expand the palette of tested olefins to the compounds containing conjugated double bond system. The premise for this is still undiminished interest in silsesquioxanes derivatives, their limited commercial availability and consequently high prices. Therefore, it seems reasonable to perform studies aimed at optimizing and modifying this group of organosilicon compounds.In studies presented in this paper, we focused on the functionalization of two types of double-decker silsesquioxane derivatives containing methyl (1A) or phenyl (1B) substituents located at 9 and 19 silicon atom of the core. Both substrates were prepared by a corner-capping reaction of incompletely condensed silsesquioxane DDSQ-4OH with vinyldichloro(organo)silane. This synthetic procedure was thoroughly described in our last paper.11b In view of the fact that methyl-substituted silicon derivatives are not reactive in metathetic transformation, DDSQ-2SiVi (1B) bearing phenyl substituents were applied to functionalization based on cross-metathesis reaction, while DDSQ-2SiVi (1A) with methyl groups was tested in silylative coupling reaction (Scheme 3).
 | ||
| Scheme 3 Silylative coupling and cross-metathesis of divinyl-substituted double-decker silsesquioxanes (1A, 1B) with 4-styryl substituted derivatives. | ||
Cross-metathesis of phenyl-substituted divinylsilsesquioxane (1B) with 1-(4-vinylphenyl)naphthalene was chosen as a model reaction for optimization studies of silsesquioxane containing two vinyl groups. Treatment of divinysilsesquioxane (1B) with two equivalents of 1-(4-vinylphenyl)naphthalene in the presence of Grubbs catalyst [RuCl2(PCy3)2(CHPh)] (1% mol in relation to vinyl group of DDSQ-2SiVi (1B) in refluxing dichloromethane, resulted in its efficient transformation. Due to the large molecular mass of the silsesquioxyl substrate (1B), the reaction progress could not be monitored by GC. After 24 h, the reaction mixture was subjected to the 1H NMR analysis which indicated nearly complete conversion of both substrates revealing selective formation of expected product (2). Similar reaction completion was observed when other 4-styryl substituted derivatives and 1B were used as reaction partners. The results obtained were collected in Table 1. In each case, the reaction led to efficient and selective formation of E-isomer of the desired product. All obtained products (1–4) are air-stable, easy to handle white or yellow solids and can be purified by column chromatography.
| DDSQ-2SiVi | R′ | Prod. no. | Isolated yield [%] |
E/Z:E/Z |
|---|---|---|---|---|
|
a Reaction conditions: CH2Cl2, reflux, [1]:[olefin] = 1:2, [RuCl2(PCy3)2(CHPh)] (2 mol%), t = 24 h, argon.
|
||||
| 1B |
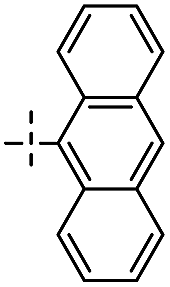
|
1 | 88 |
E:E |
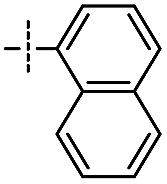
|
2 | 90 | ||
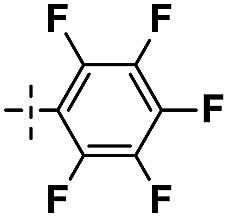
|
3 | 92 | ||
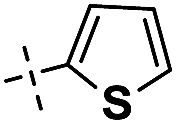
|
4 | 89 | ||
In the next experiments, silylative coupling of methyl-substituted divinylsilsesquioxane (1A) with 4-styryl substituted derivatives was performed. A series of catalytic tests aimed at optimizing the reaction systems for efficient and selective synthesis of dialkenyl-substituted derivatives (5–8) was carried out. Optimization involved selection of a suitable solvent, ratios of reactants, concentration of catalyst used, reaction temperature and time as well as determination of possible influence of cocatalyst on the reaction course. According to obtained results of performed tests, effective functionalization of 1A based on silylative coupling may be conducted in two ways concerning different reaction conditions. The first reaction was carried out in toluene at 90 °C or 110 °C under constant flow of argon in the presence of ruthenium(II) hydride complex [RuHCl(CO)(PCy3)2] used at 2 mol% (1% mol in relation to vinyl group of DDSQ-2SiVi (1A). Similar effect can be achieved by introducing a cocatalyst to the reaction system which is copper(I) chloride. The addition of the afore-mentioned salt helps to reduce the reaction temperature to 45 °C and change the solvent from toluene to methylene chloride. The results obtained are collected in Table 2.
| DDSQ-2SiVi | R′ | Prod. no. | Isolated yield [%] |
E/Z:E/Z |
|
|---|---|---|---|---|---|
| System 1 | System 2 | ||||
|
a Reaction conditions: system 1: toluene, 90 °C, [1]:[olefin] = 1:3, [RuHCl(CO)(PCy3)2] (2 mol%), t = 24 h, argon; * 110 °C, t = 96 h. System 2: CH2Cl2, 45°, [1]:[olefin] = 1:2, [RuHCl(CO)(PCy3)2] (2 mol%), [Ru]:[CuCl] = 1:5, t = 24 h, argon.
|
|||||
| 1A |

|
5 | 72 | 85 |
E:E |
|
6 | 81 | 90 | ||

|
7 | 86 | 91 | ||

|
8 | 62* | 89 | ||
The mechanism explaining the effect of copper(I) chloride addition on the reaction course was not examined. A reasonable explanation reported in literature for similar systems indicates that copper salt acts as a phosphine scavenger and thus facilitates the formation of catalytically active species.14,15 Both reaction systems were tested using different 4-styryl substituted derivatives and in each case complete conversion of silsesquioxyl substrate 1A was achieved.
The results presented in Table 2 show that irrespective of the type of olefin used, all reactions are stereoselective and lead exclusively to the product of E geometry of the resulting substituted vinylene bond (1H NMR data along with the high values of JHH coupling constant are to confirm this). The reactions performed with copper chloride(I) allow the preparation of the desired products under mild reaction conditions. However, it is necessary to isolate the 4-styryl substituted derivatives of 1A from the reaction mixtures on chromatographic columns. This necessity follows mainly from poor solubility of the copper salt used in the solvent that precipitates silsesquioxyl products (methanol, acetone, n-hexane). Furthermore, these systems may be applied only in the case of olefins soluble in refluxing methylene chloride. Applying harsh conditions, i.e. toluene, 90 °C or 110 °C, promotes (or increases) the homogeneity of the system as with increasing temperature the solubility of most styryl derivatives also increases. Therefore, these systems can be advantageously used for substrates that cause solubility problems. Both developed reaction systems are characterized by high stereoselectivity and selection of appropriate conditions for functionalization largely depend on the type olefins used.
Synthesis of macromolecular compounds with DDSQ embedded in the linear chain – ADMET and silylative coupling copolycondensation
Positive results obtained for the functionalization of divinylsilsesquioxanes via cross-metathesis and silylative coupling with a series of para-substituted styrenes with highly π-conjugated substituents prompted us to extend the range of olefins to diolefins. According to literature processes, we synthesized a number of distyrylarenes, which were then subjected to SCC and ADMET copolymerization with DDSQ-2SiVi. The overview of this processes is presented in Scheme 4.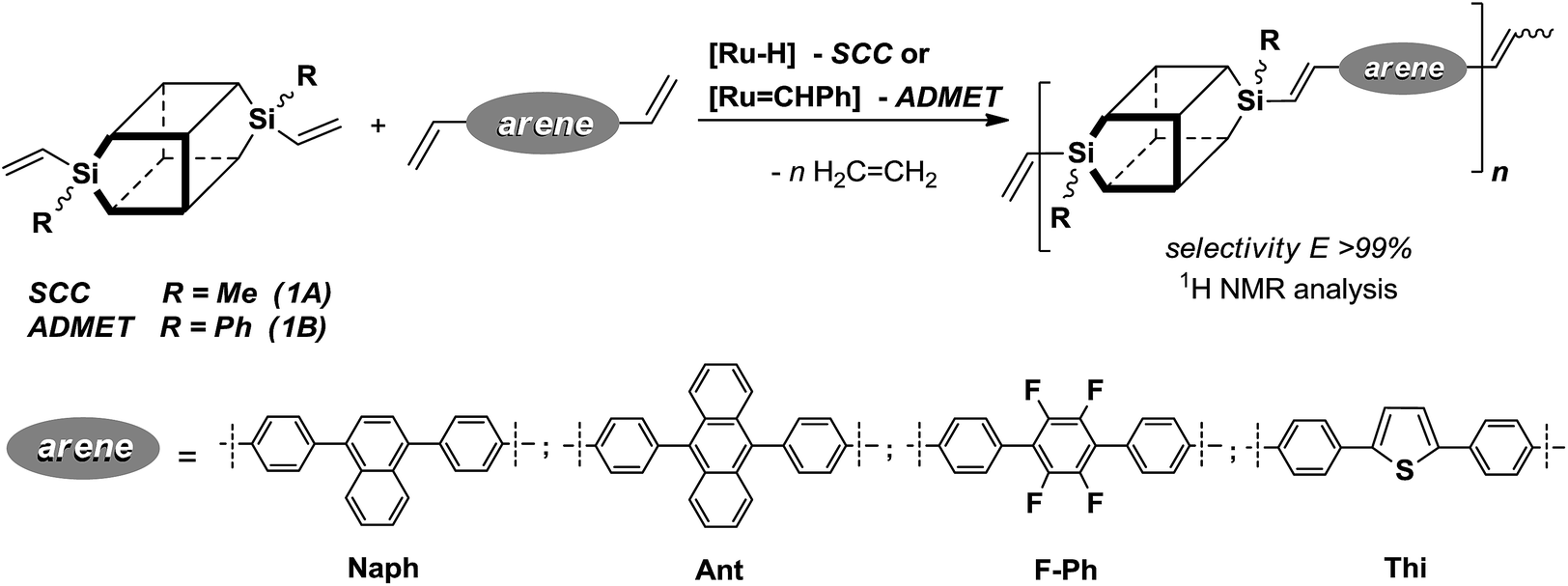 | ||
| Scheme 4 Silylative coupling copolycondensation (SCC) and metathetic copolymerisation (ADMET) of divinyl-substituted double-decker silsesquioxanes (1A, 1B) with diolefins. | ||
As a result of a series of catalytic tests, successful procedures that ensure the efficiency of both reactions were developed. They allow avoidance of generation of the undesirable polyarenes without the need for any inhibitor addition. Analogously, as in the synthesis of the molecular products, for the studies on metathetic copolymerization (ADMET), phenyl-substituted silsesquioxane (1B) was applied exclusively whereas in the silylative coupling copolycondensation double-decker silsesquioxane derivative with methyl group at 9 and 19 silicon atom (1A) was used. After optimization of the reaction conditions, a series of copolymers of stereoregular DDSQ-silylene-vinylene-arylene units was obtained (Table 3). As indicated in Table 3, metathetic transformation failed to give cooligomer containing sulfur atom in its structure (16). This may be associated with the lack of solubility of the distyrylthiophene (Thi) used in refluxing methylene chloride. There was also no reaction progress observed when increasing the amount of the solvent used or when twofold load of alkylidene catalyst was used. To the best of our knowledge, no experiments with Grubbs I generation catalyst have been conducted at high temperatures because of its thermal degradation and in situ formation of ruthenium hydride species that catalyze silylative coupling process.16 Co-oligomer containing thiophene derivative was synthesized only by SCC in the high temperature system ensuring the solubility of the diolefin used.
| DDSQ-2SiVi | R′ | Prod. no. | SCC isolated yield [%] | ADMET isolated yield [%] | |
|---|---|---|---|---|---|
| System 1 | System 2 | ||||
|
a Reaction condition: SCC reaction: system 1: toluene, 110 °C; [1]:[diolefin] = 1:1; RuHCl(CO)(PCy3)2 (2 mol%), t = 72 h, argon; system 2: CH2Cl2, 45 °C, [1]:[diolefin] = 1:1, [RuHCl(CO)(PCy3)2] (2 mol%), [Ru]:[CuCl] = 1:5, t = 72 h, argon. ADMET reaction: CH2Cl2, reflux, argon, [1]:[diolefin] = 1:1, [RuCl2(PCy3)2(CHPh)] (2 mol%), t = 72 h.
|
|||||
| 1A |
|
9 | 93 | 89 | — |
| 1B | 10 | — | — | 90 | |
| 1A |

|
11 | 90 | 86 | — |
| 1B | 12 | — | — | 85 | |
| 1A |
|
13 | 91 | 85 | — |
| 1B | 14 | — | — | 93 | |
| 1A |
|
15 | 80 | 0 | — |
| 1B | 16 | — | — | 0 | |
The new oligomeric products obtained were purified by twice repeated precipitation in methanol or n-hexane. Products obtained in the presence of catalytic systems containing copper chloride(I) required further purification on column chromatography with silica gel because the co-catalyst also was not soluble in the precipitant solvent. All oligomeric materials are white or yellow solids and show good solubility in common organic solvents, e.g. THF, CHCl3 and CH2Cl2. They were analyzed by spectroscopic methods.
Subsequently, all prepared samples were analyzed by gel permeation chromatography. They were also subjected to thermogravimetric analysis to evaluate the effect of the co-monomers used and the type of catalytic process on their molecular weights, molecular weight distribution and thermal stability. GPC results obtained for cooligomers synthesized via silylative coupling copolycondensation (SCC) and ADMET copolymerization processes are summarized in Table 4.
| DDSQ-2SiVi | R′ | Prod. no. | M n | M w | PDI | Purity% |
|---|---|---|---|---|---|---|
| 1A |
|
9 | 5200 | 7300 | 1.4 | 100 |
| 1B | 10 | 9100 | 13600 |
1.5 | 91 | |
| 1A |

|
11 | 6000 | 8900 | 1.5 | 100 |
| 1B | 12 | 13000 |
29200 |
2.2 | 95 | |
| 1A |

|
13 | 5400 | 6000 | 1.1 | 46 |
| 1B | 14 | 18300 |
46100 |
2.5 | 98 | |
| 1A |
|
15 | 5200 | 11000 |
2.1 | 61 |
| 1B | 16 | — | — | — | — |
On the basis of the data obtained, it can be noted that ADMET process led to formation of products of relatively higher molecular weights (Mn and Mw) and higher PDI values except for co-oligomer containing thiophene derivative. These results are in good agreement with those in the few available papers concerning the synthesis of similar linear polymers with a DDSQ spacer and π-conjugated arenes. Kakimoto et al. obtained analogous systems with Mn weight in the range of 11900 to 29100, and Mw/Mn values of 2.9–4.9.3e
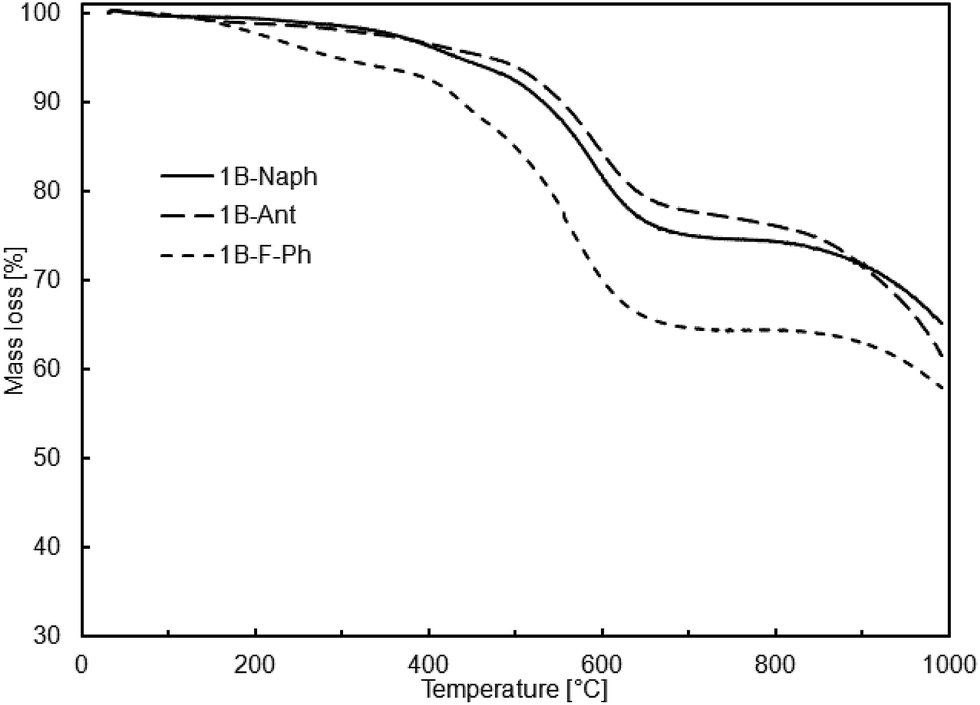
According to the thermogravimetric studies, all prepared cooligomers show relatively high thermal stability (Fig. 1 and 2).
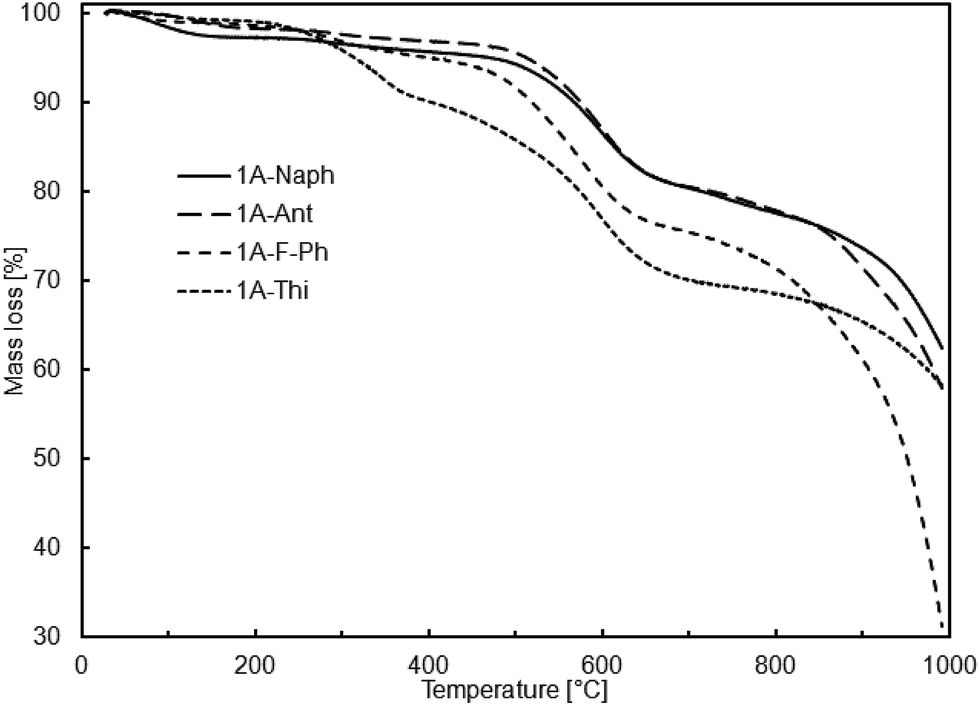 | ||
| Fig. 1 TGA curves of obtained cooligomers viaSCC. | ||
 | ||
| Fig. 2 TGA curves of obtained cooligomers viaADMET copolymerization. | ||
It is worth noting that there is relatively good correlation between the sample thermal stability and its purity or more precisely, the content of higher molecular weight fraction. It may be an explanation why the samples obtained viaSCC process are more thermally stable despite their relatively lower molecular weights in comparison to the samples obtained viaADMET copolymerization (Tables 4 and 5).
| DDSQ-2SiVi | R′ | Prod. no. | Temp. of mass loss [°C] | Residue at 1000 °C [%] | |
|---|---|---|---|---|---|
| 5% | 10% | ||||
| 1A |
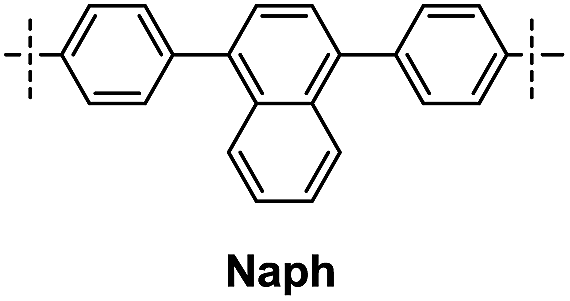
|
9 | 464 | 567 | 62 |
| 1B | 10 | 431 | 531 | 65 | |
| 1A |

|
11 | 512 | 575 | 58 |
| 1B | 12 | 467 | 553 | 62 | |
| 1A |
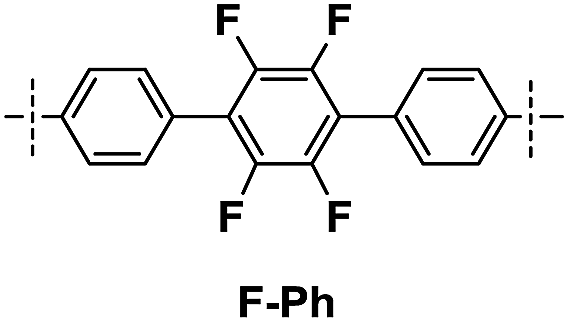
|
13 | 398 | 518 | 31 |
| 1B | 14 | 291 | 437 | 58 | |
| 1A |
|
15 | 314 | 403 | 58 |
| 1B | 16 | — | — | — | |
So far, there has been scarce information on similar oligomeric systems with DDSQ fragments. However, among those rare examples in literature, high temperatures of 10% mass loss, reaching over 550 °C, are characteristic of DDSQ copolymers irrespective of the comonomer or catalytic process used.3h This effect may be also attributed to the general tendency of the high thermal stability of POSS compounds heaving phenyl groups.3e
Conclusions
Recent years have shown that the concept of synthesis of molecular and macromolecular organosilicon compounds based on DDSQ led to two major trends in their development. Molecular derivatives based on a DDSQ core are a class of compounds that may be synthesized using, among others, stoichiometric types of modifications, i.e. a condensation of tetrasilanolphenyl POSS (DDSQ-4OH) with alkoxy- or chlorosilanes. On the other hand, difunctional DDSQ compounds with reactive groups, e.g. a Si–H or Si–HCCH2 bond, are ideal substrates for further catalytic modifications. We described divinyl-substituted double-decker silsesquioxanes that undergo efficient functionalization via cross-metathesis and silylative coupling reactions with a series of para-substituted styrenes that possess highly π-conjugated substituents. For all styrene derivatives tested, both reactions proceeded effectively and with high stereoselectivity leading to exclusive formation of E isomer. A divinyl-substituted double-decker silsesquioxane containing a methyl group located at Si-9 and Si-19 atoms (1A) was inactive in metathetic transformation, but this result was expected in the context of our earlier reports.17 Cross-metathesis and silylative coupling silylative are convenient and complementary synthetic routes leading to functionalization of vinyl-substituted silicon derivatives. However, the choice of the optimum catalytic method depends on the specific character of the reaction system. The other synthetic path for DDSQ applications is to try to embed them into the main chain of the polymeric system. We proved that both, SCC and ADMET reactions may be used for the stereoselective synthesis of a new class of vinylene-arylene copolymers containing DDSQ in the main chain. New copolymeric material was characterized in terms of molecular weights, PDI as well as thermal analysis. TG analysis has proved the high level of thermal resistance of the obtained systems, reaching over 550 °C, that is in close correlation with DDSQ copolymers.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Science Centre (Poland) (MAESTRO Project No. DEC-2011/02/A/ST5/00472, SONATA Project No. DEC-2012/05/D/ST5/03348).Notes and references
- (a) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed; (b) D. B. Cordes and P. D. Lickiss, in Applications of Polyhedral Oligomeric Silsesquioxanes, Advances in Silicon Science 3, ed. C. Hartmann-Thompson, Springer, 2011 Search PubMed.
- (a) Y. Morimoto, K. Watanabe, N. Ootake, J. Inagaki, K. Yoshida and K. Ohguma, EP 1 428 795 (A1), 2004; US 7 169 873 (B2), 2007; US 7 449 539 (B2), 2008; (b) K. Yoshida, Polymer Preprints Japan, 2003, 52, 316 Search PubMed; (c) N. Ootake and K. Yoshida, US pat., 2006/0155091(A1), 2006.
- (a) A. C. Kucuk, J. Matsui and T. Miyashita, Thin Solid Films, 2013, 534, 577 CrossRef CAS; (b) B. Seurer, V. Vij, T. Haddad, J. M. Mabry and A. Lee, Macromolecules, 2010, 43, 9337 CrossRef CAS; (c) M. Kohri, J. Matsui, A. Watanabe and T. Miyashita, Chem. Lett., 2010, 39, 1162 CrossRef CAS; (d) J. Espinas, J. D. A. Pelletier, E. Abou-Hamad, L. Emsley and J.-M. A. Basset, Organometallics, 2012, 31, 7610 CrossRef CAS; (e) M. Seino, T. Hayakawa, Y. Ishida, M. Kakimoto, K. Watanabe and H. Oikawa, Macromolecules, 2006, 39, 3473 CrossRef CAS; (f) K. Yoshida, T. Hattori, N. Ootake, R. Tanaka and H. Matsumoto, Silsesqioxane-based polymers: synthesis of phenyl silsesquioxanes with double-decker structure and their polymers, in Silicon Based Polymers. Advances in Synthesis and Supramolecular Organization, ed. F. Ganachaud, S. Boileau and B. Boury, Springer, London-New York, 2008, p. 205 Search PubMed; (g) S. Wu, T. Hayakawa, M. Kakimoto and H. Oikawa, Macromolecules, 2008, 41, 3481 CrossRef CAS; (h) M. Miyasaka, Y. Fujiwara, H. Kudo and T. Nishikubo, Polym. J., 2010, 42, 799 CrossRef CAS; (i) K. Wei, L. Wang and S. Zheng, Polym. Chem., 2013, 4, 1491 RSC; (j) K. Wei, L. Wang and S. Zheng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4221 CrossRef CAS; (k) W. Zhang, J. Xu, X. Li, G. Song and J. Mu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 780 CrossRef CAS; (l) K. Wei, L. Wang, L. Li and S. Zheng, Polym. Chem., 2015, 6, 256 RSC; (m) B. Dudziec and B. Marciniec, Curr. Org. Chem., 2015 DOI:10.2174/1385272820666151228193728.
- (a) B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374 CrossRef CAS; (b) B. Marciniec, Acc. Chem. Res., 2007, 40, 943 CrossRef CAS PubMed; (c) B. Marciniec and C. Pietraszuk, in Green Metathesis Chemistry. Great Challenges in Synthesis Catalysis and Nanotechnology, ed. V. Dragutan, A. Demonceau, I. Dragutan and E. S. Finkelshtein, Springer, Berlin, 2010, p. 157 Search PubMed; (d) C. Pietraszuk, P. Pawluć and B. Marciniec, in Handbook on Metathesis, R. Grubbs and D. J. O'Leary, 2015, vol. 2, 9, pp. 583–631 Search PubMed.
- (a) Y. Itami, B. Marciniec and M. Kubicki, Chem.–Eur. J., 2004, 10, 1239 CrossRef CAS PubMed; (b) J. Waehner, B. Marciniec and P. Pawluc, Eur. J. Inorg. Chem., 2007, 2975 CrossRef CAS; (c) P. Żak, C. Pietraszuk, B. Marciniec, G. Spolnik and W. Danikiewicz, Adv. Synth. Catal., 2009, 351, 2675 CrossRef; (d) P. Żak, B. Marciniec, M. Majchrzak and C. Pietraszuk, J. Organomet. Chem., 2011, 696, 887 CrossRef.
- F. J. Feher, D. Soulivong, A. G. Eklund and K. D. Wydham, Chem. Commun., 1997, 1185 RSC.
- S. Sulaiman, A. Bhaskar, J. Zhang, R. Guda, T. Goodson III and R. M. Laine, Chem. Mater., 2008, 20, 5563 CrossRef CAS.
- (a) G. Cheng, N. R. Vautravers, R. E. Morris and D. J. Cole-Hamilton, Org. Biomol. Chem., 2008, 6, 4662 RSC; (b) N. R. Vautravers, P. Andre, A. M. Z. Slavin and D. J. Cole-Hamilton, Org. Biomol. Chem., 2009, 7, 717 RSC; (c) N. R. Vautravers, P. Andre and D. J. Cole-Hamilton, J. Mater. Chem., 2009, 19, 4545 RSC.
- H. Araki and K. Naka, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4170 CrossRef CAS.
- M. Lo Conte, S. Staderini, A. Chambery, N. Berthet, P. Dumy, O. Renaudet, A. Marra and A. Dondoni, Org. Biomol. Chem., 2012, 10, 3269 CAS.
- (a) P. Żak, B. Dudziec and B. Marciniec, US Appl. US 14,640,618, EP 15461513.2, 2015; (b) P. Żak, B. Dudziec, M. Kubicki and B. Marciniec, Chem.–Eur. J., 2014, 20, 9387 CrossRef PubMed.
- (a) M. Majchrzak, S. Kostera, M. Kubicki and I. Kownacki, Dalton Trans., 2013, 42, 15535 RSC; (b) M. Majchrzak, M. Hybsz, S. Kostera, M. Kubicki and B. Marciniec, Tetrahedron Lett., 2014, 55, 3055 CrossRef CAS.
- C. S. Yi, D. W. Lee and Y. Chen, Organometallics, 1999, 18, 2043 CrossRef CAS.
- P. Żak, M. Kubicki, B. Marciniec, S. Rogalski, C. Pietraszuk and D. Frąckowiak, Dalton Trans., 2014, 43, 7911 RSC.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887 CrossRef CAS.
- B. Schmidt, Eur. J. Org. Chem., 2004, 1865 CrossRef CAS.
- C. Pietraszuk and H. Fischer, Chem. Commun., 2000, 2463 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20848c |
| This journal is © The Royal Society of Chemistry 2016 |